
Varför är insulin unikt? #
De flesta biologiska kontrollsystem, inklusive fysiologiska mekanismer, kännetecknas av inbyggd redundans som säkerställer att ett system kan kompensera för ett annat när det fallerar. Ett exempel är att flera hormoner kan höja blodsockernivåerna genom olika mekanismer.
Insulin är dock det enda hormonet som har förmågan att sänka blodsockret. Vid första anblick kan detta framstå som oväntat, men det är avgörande att förstå att ett överskott av insulin kan ha långt mer akuta och förödande konsekvenser än en brist.
Om blodsockret sjunker under 2 mmol/l under en så kort period som fem minuter kan det leda till irreversibel hjärnskada och i värsta fall vara livshotande. Däremot tar det ofta månader eller till och med år av kroniskt förhöjda blodsockernivåer, till följd av långvarig insulinbrist, innan de allvarliga komplikationerna av diabetes manifesterar sig.
Insulin kan därför betraktas som ett hormon där både överproduktion och underproduktion är förenade med stora risker. Även om insulinbrist och utvecklingen av diabetes får omfattande uppmärksamhet, är det viktigt att understryka att en akut insulinöverdos ofta har betydligt allvarligare konsekvenser.
En annan funktion av insulin är dess roll som tillväxtstimulerande hormon, en egenskap det delar med andra hormoner som insulinliknande tillväxtfaktor 1 och 2 (IGF-1 och IGF-2). Däremot är insulins unika roll i att reglera glukoshomeostasen utan motsvarighet i andra hormonsystem, vilket innebär att det saknar redundans i denna funktion.
Ett evolutionärt persepektiv
Det finns spekulationer bland fysiologer att den potentiella faran med hypoglykemi är anledningen till att insulin, via en enda receptor, ensam reglerar sänkningen av blodsockret. Ur ett evolutionärt perspektiv var hypoglykemi, snarare än hyperglykemi, den största risken under människans tidiga historia.
Perioder av begränsad tillgång till föda och intensiv fysisk aktivitet, som exempelvis vid flykt från rovdjur, innebar att ett system för att höja blodsockret var avgörande, medan mekanismer för att sänka blodsockret behövde vara strikt reglerade för att undvika livshotande hypoglykemi.
I detta sammanhang var det evolutionärt fördelaktigt att ha flera redundanta system för att höja blodsockret, medan endast ett hormon, insulin, utvecklades för att sänka det. Denna balans säkerställde att risken för oavsiktlig hypoglykemi hölls till ett minimum.
β-celler och insulinfrisättning #
β-celler har utvecklat specifika metabola mekanismer för att förhindra överdriven insulinfrisättning och därmed hypoglykemi, särskilt under fysisk aktivitet.
För det första är insulinfrisättningen ytterst känslig för förändringar i blodsockernivåer. Detta uppnås genom att koppla glukosmetabolism till insulinfrisättning via förändringar i intracellulära ATP-nivåer, β-cellernas elektriska aktivitet och frisättningen av insulinvesiklar.
Oxidativ fosforylering
Detta innebär att när blodsockernivån i kroppen ökar, tas glukos upp av β-cellerna i bukspottkörteln, som är ansvariga för att producera och frisätta insulin. Majoriteten av detta glukos metaboliseras (bryts ner för att skapa energi) genom en process som kallas oxidativ fosforylering.
Oxidativ fosforylering i β-cellerna:
1. Glukos tas upp: β-celler tar upp glukos från blodet via specifika transportproteiner, till exempel GLUT2.
2. Metabolism: Inuti β-cellerna bryts glukos ner genom glykolys och citronsyracykeln, vilket leder till produktion av energirika molekyler, såsom NADH och FADH2.
3. Elektrontransportkedjan: Dessa energirika molekyler används i mitokondrierna för att driva elektrontransportkedjan, en del av oxidativ fosforylering.
4. ATP-produktion: Slutprodukten är ATP (adenosintrifosfat), en viktig energikälla för cellerna.
I β-celler är ATP centralt för att reglera frisättningen av insulin. När nivåerna av ATP ökar:
- Blockeras ATP-beroende kaliumkanaler.
- Detta leder till en förändring i cellens elektriska laddning.
- Depolariseringen öppnar spänningsstyrda kalciumkanaler, vilket gör att kalcium strömmar in i cellen.
- Den ökade kalciumnivån stimulerar exocytos, vilket innebär att insulin frisätts från β-cellerna till blodet.
Omvänt, när blodsockernivåerna sjunker, upphör insulinfrisättningen snabbt. Detta beror på en minskning av intracellulärt ATP, vilket öppnar KATP-kanalerna, leder till hyperpolarisering av membranet, minskat kalciuminflöde och därigenom en inhibering av insulinfrisättningen.
Vad orsakar insulinbristen vid typ 2 diabetes? #
Den cellulära sekretoriska kapaciteten och β-cellmassa
Den nedsatta insulinsekretionen vid T2DM kan förklaras av en minskning i β-cellernas individuella sekretoriska funktion, en reduktion av β-cellmassan (vilket är produkten av β-cellernas storlek och antal), eller en kombination av dessa faktorer.
Trots omfattande forskning och långvarig debatt kring betydelsen av sekretorisk dysfunktion jämfört med förlust av β-cellmassa för nedsatt insulinfrisättning vid T2DM saknas fortfarande en enhetlig konsensus.
En av de huvudsakliga utmaningarna i detta sammanhang är svårigheten att erhålla tillräckligt högkvalitativa och kvantitativa prover från isolerade Langerhanska öar, särskilt från donatorer med T2DM, för funktionella studier.
Variabler som påverkar jämförande studier
Flera faktorer komplicerar jämförelser mellan funktion hos kontroll- och T2DM-öar. Exempelvis kan donatorer ha varit under behandling med olika läkemedelskombinationer före döden, vilket kan påverka β-cellernas funktion. Vidare kan genetiska och miljömässiga faktorer vara dåligt kontrollerade.
Dessutom kan skillnader i kall ischemitid, den tid pankreas exponeras för låga temperaturer under transport och isolering, påverka genuttryck och funktion i de isolerade öarna. Dessa variabler begränsar den övergripande tillförlitligheten i resultaten från sådana studier.
β-cellmassa och histologiska studier
Histologisk analys av β-cellmassa
Histologiska studier är enklare att genomföra eftersom de kan utföras på fixerade vävnadsprover. Flera studier har rapporterat en minskning av β-cellmassan hos patienter med T2DM. En viktig begränsning med dessa analyser är dock att β-celler oftast identifieras genom immunfärgning av insulin.
Detta innebär att β-celler med starkt reducerat insulininnehåll inte detekteras, vilket kan leda till en underskattning av β-cellmassan.
Upptäckter med avancerad teknik
Nya studier som använder elektronmikroskopi har avslöjat att β-celler i T2DM-öar kan identifieras baserat på deras unika “pocherade ägg”-utseende hos insulingranula. Trots att dessa granula är få och insulin är odetekterbart med konventionell immunfärgning, kvarstår cellerna.
Liknande fynd har observerats i musmodeller med diabetes, där hyperglykemi orsakar en minskning av insulinhalten i β-celler utan att nödvändigtvis minska det totala antalet celler.
Osäkerheter och kvantifiering av β-cellförlust
Även om det är tydligt att insulinhalten i β-celler minskar med tiden, är omfattningen av den faktiska förlusten av β-cellmassa fortfarande oklar. Trots detta finns robust evidens för att β-cellmassan hos patienter med T2DM minskar med cirka 25 % under de första fem åren efter diagnos jämfört med icke-diabetiska kontroller. Vid långvarig T2DM (>15 år) är β-cellmassan reducerad med över 50 %.
Sekretorisk belastning och β-cellers motståndskraft
Den gradvisa förlusten av β-cellmassa under sjukdomens utveckling leder till en allt större sekretorisk belastning på de kvarvarande funktionella β-cellerna. Dessa cellers motståndskraft och funktionalitet bestäms av ett komplext samspel mellan miljöfaktorer, genetiska predispositioner och epigenetiska mekanismer.
Att förstå detta samspel är avgörande för att utveckla nya terapier som kan bromsa eller reversera β-cellförlusten och förbättra den sekretoriska kapaciteten vid T2DM.
β-cellers dedifferentiering
Det är tydligt att flera mekanismer bidrar till utvecklingen av T2DM. Ny forskning indikerar dock att β-cellernas identitet är dynamisk och att förändringar i denna kan bidra till defekt insulinsekretion vid T2DM.
Hyperglykemi hos möss leder till förändrat uttryck av β-cell-specifika transkriptionsfaktorer, vilket orsakar dedifferentiering och nedsatt insulinsekretion. Studier har visat att deletion av transkriptionsfaktorer som FOXO1 kan leda till att β-celler förlorar sitt insulininnehåll och återgår till en progenitorliknande celltyp.
På liknande sätt har uttryck av progenitormarkören Ngn3 observerats i musmodeller för diabetes. Huruvida liknande dedifferentiering sker hos humana β-celler är ännu oklart, men förändringar i β-cell-transkriptionsfaktorer har dokumenterats hos både människor med T2DM och primater med kostinducerad prediabetes.
Förlusten av insulinuttryck i musmodeller följs ofta av en ökning i glukagonuttryck, sannolikt inducerad av hyperglykemi. Linjespårning har visat att vissa β-celler börjar uttrycka glukagon, men det är oklart om dessa helt omvandlas till α-celler eller representerar en intermediär celltyp som uttrycker både glukagon och β-cellproteiner (utom insulin). Samtidigt har α- och δ-celler i vissa fall visats kunna omvandlas till fungerande β-celler, vilket betonar isletcellernas plasticitet.
Mekanismer bakom β-cellsdysfunktion #
Mekanismer bakom β-cellsdysfunktion vid T2DM
Komplexa interaktioner och cellulär stress
Traditionellt har β-cellsdysfunktion vid T2DM kopplats till β-cellsdöd, men ny forskning visar att den snarare är resultatet av komplexa interaktioner mellan miljöfaktorer och molekylära mekanismer. Vid fetma och näringsöverskott uppstår ofta hyperglykemi, hyperlipidemi, insulinresistens och kronisk inflammation. Dessa tillstånd utsätter β-celler för toxisk stress som inflammation, ER-stress, oxidativ stress och amyloid stress, vilket hotar ö-cellernas integritet.
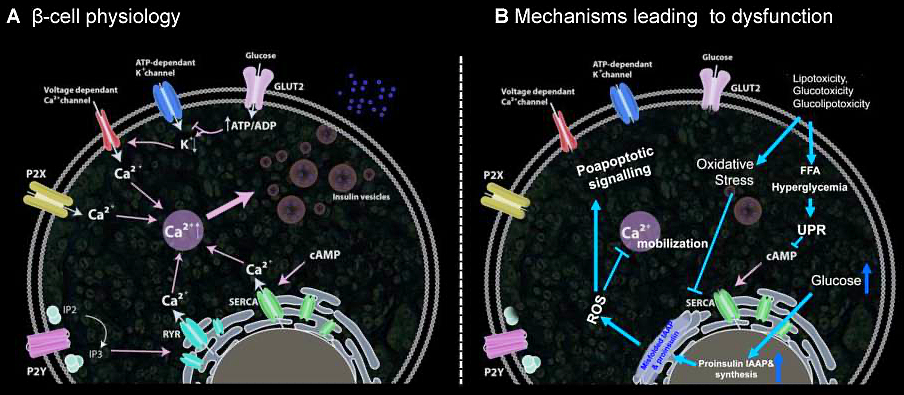
Figuren ovan visar hur beta-cellens fysiologiska svar och insulinsekretion förändras hos individer med beta-cellsdysfunktion. Det sker en successiv apoptos av beta-celler till följd av ökad ROS nivåer.
Lipotoxicitet, glukotoxicitet och ER-stress
Överskott av fria fettsyror (FFA) och hyperglykemi orsakar ER-stress genom att aktivera apoptotiska UPR-vägar. Mättade FFA hämmar SERCA (som reglerar ER-Ca²⁺-mobilisering), aktiverar IP3-receptorer och stör ER-homeostas. Samtidigt leder långvarigt högt glukos till ökad proinsulinsyntes och IAAP-produktion, vilket resulterar i ackumulering av felveckade proteiner och överproduktion av ROS.
Detta försämrar den normala ER-Ca²⁺-mobiliseringen och triggar apoptotiska signaler samt lokal inflammation via IL-1β-frisättning.
Konsekvenser för ö-cellernas integritet
Den kroniska stressen bidrar till störd organisation och kommunikation mellan cellerna i pankreasöarna. Detta påverkar insulin- och glukagonfrisättning negativt och förvärrar hyperglykemin. Minskad uttrycksnivå av GLUT2-glukostransportören eller brister i veckningen av proinsulin är exempel på mekanismer som leder till insulinsekretorisk dysfunktion, en central faktor vid T2DM.
Dynamiken i insulinfrisättning #
Den tidiga fasen av insulinfrisättning
Vid T2DM är den initiala fasen av insulinfrisättning efter måltid avsevärt nedsatt, vilket ofta observeras redan hos individer med lätt nedsatt glukostolerans. Denna tidiga fas är avgörande för att snabbt undertrycka leverns glukosproduktion efter en måltid.
När mekanismen inte fungerar leder det till förhöjda blodsockernivåer, vilket ökar belastningen på β-cellerna och resulterar i en fördröjd och överdriven insulinproduktion senare. Denna kompensatoriska insulinfrisättning, som tidigare tolkades som ett tecken på insulinresistens, speglar snarare β-cellernas anpassning till ett dysreglerat system.
Forskning har visat att återställning av den tidiga insulinfrisättningen, genom exogen insulinbehandling eller läkemedel som stimulerar β-cellerna, kan minska behovet av den sena kompensatoriska fasen. Detta bidrar till förbättrad glukoskontroll och avlastning av β-cellerna, vilket kan fördröja sjukdomsprogressionen.
Betydelsen av glukosnivåer för insulinfrisättning
Insulinfrisättning är starkt reglerad av blodsockernivåerna. Vid T2DM kan β-celler producera högre nivåer av insulin jämfört med friska individer, men detta är ofta otillräckligt för att möta de kroniskt förhöjda glukosnivåerna. Därför är det avgörande att utvärdera insulinnivåerna i förhållande till glukosnivåerna för att korrekt bedöma β-cellernas funktion.
Hyperglykemiska klamptester #
Hyperglykemiska klamptester betraktas som “guldstandarden” för att analysera β-cellernas funktion. Genom att ge en standardiserad glukosstimulering möjliggör dessa tester en detaljerad utvärdering av både den tidiga och sena fasen av insulinfrisättning.
Detta verktyg är ovärderligt för att förstå patofysiologin vid T2DM och för att identifiera mål för terapier som syftar till att bevara β-cellernas funktion och förbättra glukoskontrollen.
Typ 2 diabetes: en progressiv sjukdom #
T2DM utvecklas gradvis genom ett antal stadier där β-cellfunktion och insulinresistens spelar avgörande roller. Dessa stadier kan delas upp enligt följande:
Utvecklingsstadier av T2DM
1. Normal glukosreglering
Glukosnivåerna är normala, men genetiska faktorer och intrauterin miljö ökar individens risk för diabetes. Detta är en asymtomatisk fas där predisponerande faktorer ackumuleras.
2. Insulinresistens och minskad insulinkänslighet
Miljöfaktorer såsom ohälsosamma livsstilsval och fetma leder till insulinresistens. För att bibehålla normoglykemi kompenserar β-celler genom att öka insulinproduktionen. Denna kompensation är dock begränsad och minskar med tiden.
3. Nedsatt glukostolerans
Vid glukosbelastning eller efter måltid är β-cellernas funktion inte längre tillräcklig för att hantera de ökande kraven, vilket leder till postprandiell hyperglykemi. Detta stadium markerar övergången till prediabetes.
4. Fasta hyperglykemi och glukotoxicitet
Fortsatt insulinresistens och förlust av β-cellfunktion resulterar i förhöjda glukosnivåer även under fasta. Glukotoxicitet förvärrar β-cellsdysfunktionen, vilket påskyndar sjukdomsprogressionen.
5. Avancerad β-cellsförlust
β-celler förlorar sin förmåga att producera tillräckliga mängder insulin, vilket leder till kronisk hyperglykemi och manifest T2DM. Den progressiva förlusten av β-cellmassa gör att de återstående β-cellerna blir överbelastade, vilket förvärrar deras funktion ytterligare.
Dynamiken mellan β-cellfunktion och insulinresistens
Forskning visar att det finns ett hyperboliskt samband mellan β-cellfunktion och insulinkänslighet. När insulinkänsligheten minskar, svarar β-cellerna genom att öka insulinproduktionen för att kompensera.
Denna anpassning kan dock bli otillräcklig vid svår insulinresistens eller vid genetiska defekter i β-cellerna, vilket leder till hyperglykemi. Hos individer med fetma förbättras β-cellernas glukoskänslighet initialt för att möta de ökade kraven, men denna kompensatoriska mekanism försvagas vid långvarig insulinresistens.
Avvikelser i insulinutsöndring och β-cellsdefekter vid typ 2-diabetes
Betydelsen av insulinsekretionens dynamik
Insulinutsöndring från β-celler sker i två faser. Den första fasen, som varar i cirka 10 minuter, involverar snabb frisättning av insulin från granuler nära β-cellens membran. Detta följs av en långsammare och mer uthållig andra fas som kräver nybildning av insulin och mobilisering av granuler från intracellulära förråd.
För att frisätta insulin svarar β-celler på förhöjda glukosnivåer genom att stänga ATP-känsliga kaliumkanaler, vilket leder till depolarisering av cellmembranet och inflöde av kalcium. Den ökade intracellulära kalciumnivån driver granulerna mot cellmembranet, där de smälter samman med det och frisätter sitt innehåll.
I typ 2-diabetes (T2DM) är dessa processer ofta störda. Den första fasen av insulinutsöndring är ofta helt frånvarande, och den andra fasen är försvagad, vilket leder till ineffektiv kontroll av blodsockernivåerna.
Insulinutsöndringens mönster under dagen
I levande organismer är insulinutsöndringen inte linjär utan sker i pulser. Dessa pulser kan vara korta (var 5–15 minut) eller långsammare och större (var 80–150 minut). De korta pulserna står för en stor del av den dagliga insulinutsöndringen och är viktiga för att upprätthålla normala blodsockernivåer.
Långsammare pulser, kända som ultradiana pulser, förstärks efter måltider och följer ofta blodsockrets variationer. Hos personer med T2DM är dessa pulser ofta oregelbundna och svagare, vilket bidrar till hyperglykemi.
Avvikelser vid T2DM och IGT
Vid diagnos av T2DM är β-cellfunktionen ofta reducerad med 50 % och fortsätter att försämras oavsett behandling. Vanliga avvikelser inkluderar:
- Frånvaro av den första fasen av insulinfrisättning.
- Försvagad andra fas av insulinfrisättning vid hyperglykemi.
- Minskad insulinsekretion vid måltider eller andra icke-glukosstimuli.
- Förändringar i pulsationerna, med svagare och mindre regelbundna mönster.
- Förhöjd kvot mellan proinsulin och insulin, vilket tyder på ineffektiv omvandling av proinsulin till insulin.
Liknande avvikelser ses hos individer med nedsatt glukostolerans (IGT), men i mindre grad. Dessa avvikelser inkluderar försämrade faser av insulinfrisättning, reducerat svar på glukos och andra stimuli samt en hög proinsulin-insulin-kvot. Hos genetiskt predisponerade individer med normala glukosnivåer kan liknande mönster observeras, vilket stärker teorin att β-cellsdefekter är en primär genetisk faktor i T2DM.
Orsaker till β-cellsdysfunktion #
Glukotoxicitet
Hyperglykemi, både långvarig och akut, kan försämra β-cellernas funktion. Studier visar att förbättrad blodsockerkontroll med hjälp av insulinfrisättande läkemedel, insulinkänslighetsförbättrande medel eller insulin kan förbättra β-cellernas funktion.
Mekanismerna bakom hyperglykemiens skadliga effekter är komplexa och involverar ökad produktion av reaktiva syreföreningar (ROS), oxidativ stress som påverkar genuttryck och proteinsyntes samt ökad β-cellapoptos. Även om glukotoxicitet är en sekundär effekt, kan den påskynda nedbrytningen av β-cellfunktionen över tid.
Lipotoxicitet
Förhöjda nivåer av fria fettsyror (FFA) i blodet, vanliga hos personer med fetma och T2DM, är också kopplade till nedsatt β-cellfunktion. Studier visar att långvariga förhöjningar av FFA kan hämma β-cellernas insulinfrisättning, försämra genuttrycket för insulin och främja β-cellapoptos.
En föreslagen mekanism involverar proteinet UCP-2, som påverkar mitokondriernas ATP-produktion och därmed minskar glukosinducerad insulinfrisättning. Hos människor är det dock svårt att påvisa dessa effekter av FFA i frånvaro av hyperglykemi, vilket leder till termen ”glukolipotoxicitet”. Det är tydligt att FFA inte är den primära orsaken till T2DM, men genetiska faktorer kan öka sårbarheten för deras negativa effekter.
Fetma
Fetma är den mest betydande förvärvade riskfaktorn för T2DM, vilket främst beror på att fetma orsakar insulinresistens. Fettvävnad frisätter faktorer som kan påverka β-cellfunk jälva mekanismen genom vilken islet-amyloid-polypeptid (IAPP) påverkar β-celler i samband med typ 2-diabetes (T2DM) är fortfarande under utredning.
Studier har föreslagit att små oligomerer av IAPP kan vara giftiga för β-celler genom att inducera stress i det endoplasmatiska retiklet, vilket i sin tur kan leda till apoptos. Dessa oligomerer verkar bildas inuti β-cellen snarare än i den extracellulära miljön.
Dessutom kan de interagera med cellmembran och orsaka permeabilitet, vilket bidrar till cellulär dysfunktion och celldöd. Trots dessa mekanismer är det ännu oklart hur stor roll IAPP-oligomerer spelar i den bredare patofysiologin för T2DM hos människor.
Betydelsen av genetiska och förvärvade faktorer
T2DM är en komplex sjukdom där genetiska och miljörelaterade faktorer samverkar för att påverka β-cellfunktion och insulinresistens. Genetiska mutationer kan påverka insulinproduktion, β-cellmassans stabilitet och β-cellernas känslighet för stressfaktorer som hyperglykemi och fria fettsyror.
Samtidigt kan förvärvade faktorer såsom fetma, höga nivåer av fria fettsyror och inflammatoriska cytokiner från fettvävnad ytterligare förvärra β-cellskadan. Det är viktigt att förstå denna samverkan för att kunna identifiera individer med hög risk för att utveckla T2DM och utveckla förebyggande strategier.
Cytokiner och deras påverkan på β-cellers funktion
Cytokiner, särskilt den proinflammatoriska cytokinen interleukin-1β (IL-1β), har visat sig spela en viktig roll i β-cellernas livscykel och funktion. IL-1β är välkänd för sin roll i cytokinförmedlad β-cellsdestruktion vid typ 1-diabetes.
På senare tid har dock IL-1β också uppmärksammats för sin roll i patogenesen av typ 2-diabetes (T2DM). Studier har visat att β-celler kan producera IL-1β som svar på höga glukos- och leptinnivåer. I pankreatiska vävnadsprover från patienter med T2DM har IL-1β-producerande β-celler identifierats, vilket inte är fallet hos friska individer.
Vid låga koncentrationer av IL-1β kan cytokinen stimulera β-cellernas proliferation och minska apoptos, men vid högre koncentrationer påverkas insulinfrisättningen negativt och apoptosen ökar. Denna apoptos medieras sannolikt via Fas, en receptor som tillhör tumörnekrosfaktorreceptorfamiljen och som kan inducera apoptotisk celldöd oberoende av TNF-α. IL-1β förstärker Fas-uttrycket i β-celler, vilket möjliggör Fas-inducerad apoptos.
Ett läkemedel som blockerar IL-1-receptorer (IL-1Ra) har visat sig skydda β-celler från IL-1β:s negativa effekter i laboratoriestudier och förbättrade även β-cellernas sekretoriska funktion hos patienter med T2DM i en preliminär studie. Trots att förhöjda nivåer av cytokiner och andra inflammatoriska förändringar är vanliga hos personer med försämrad glukostolerans och T2DM, är deras exakta bidrag till β-cellsdysfunktion fortfarande oklart.
Förändringar i Langerhanska öar och β-celler vid typ 2 diabetes #
De Langerhanska öarna i bukspottkörteln utgör 3–5 % av den vuxna pankreasvävnaden, och β-celler står för 60–80 % av cellerna i dessa öar. Vid T2DM förekommer flera histologiska förändringar i öarna, inklusive amyloidinlagringar, minskad β-cellmassa och förändringar i cellpopulationens proportioner.
Histologiska förändringar i öarna
Ett utmärkande patologiskt kännetecken hos 90 % av patienter med T2DM är förekomsten av amyloidinlagringar i Langerhanska öarna. Dessa inlagringar består av islet amyloid polypeptid (IAPP), även kallad amylin, som är en peptid med 37 aminosyror. IAPP produceras normalt av β-celler, lagras tillsammans med insulin i sekretoriska vesiklar och frisätts tillsammans med insulin som svar på stimuli. Vid T2DM genomgår lösliga IAPP-peptider en konformationsändring och bildar olösliga amyloidinlagringar.
Det är fortfarande oklart vad som initierar denna process, men det spekuleras att den reflekterar en försämring och destruktion av β-celler. Liknande amyloidinlagringar kan också förekomma hos en mindre andel äldre individer utan diabetes. Nyare forskning pekar på att små intracellulära oligomerer av IAPP snarare än amyloidfibriller är de cytotoxiska former som kan kopplas till ökad β-cellapoptos i djurstudier.
Förändringar i β-celler
β-cellmassan hos personer med T2DM är generellt minskad med cirka 30–50 % jämfört med viktmatchade individer med normal glukostolerans. Denna minskning har också observerats hos personer med försämrat fasteglukos.
Minskningen av β-cellmassan beror främst på en minskning i antalet celler snarare än deras storlek. En markant ökning av β-cellapoptos, utan tillräcklig kompensation genom celldelning eller nybildning, är den primära orsaken till minskningen av antalet β-celler.
Vidare har en minskning av antalet sekretoriska vesiklar i β-cellerna rapporterats, vilket ytterligare bidrar till försämrad insulinfrisättning. Fibros i Langerhanska öarna och omgivande endokrin vävnad är också en vanlig observation vid T2DM. Dessa förändringar återspeglar sjukdomens komplexa natur och utgör en central del av patogenesen vid T2DM.
Förändringar i β-, α- och δ-celler vid typ 2 diabetes
β-cellförändringar och insulinproduktion
Den reducerade β-cellmassan vid typ 2-diabetes (T2DM) är inte ensam tillräcklig för att förklara den minskade insulinsekretionsförmågan. Detta indikerar att det finns funktionella defekter som potentiellt är reversibla. Trots en minskning i β-cellernas antal finns det möjligheter för återställning av vissa funktioner genom att adressera underliggande faktorer som glukotoxicitet och lipotoxicitet.
α-cellförändringar och glukagonproduktion
Vid T2DM är α-cellmassan antingen oförändrad eller något ökad, vilket leder till en relativ ökning av α-cellandelen eftersom β-cellmassan minskar. Abnorm α-cellfunktion vid försämrad glukostolerans (IGT) och T2DM inkluderar:
- Försämrad hämning av hyperglykemi.
- Överdriven respons på aminosyror eller blandade måltider.
- Minskad respons vid hypoglykemi.
Dessa förändringar har korrelerats med reducerad β-cellfunktion och kan därför vara sekundära fenomen som påverkas av glukotoxicitet, lipotoxicitet och insulinresistens. Hyperglukagonemi, som innebär förhöjda nivåer av glukagon, är ett kännetecken för både typ 1- och typ 2-diabetes. Detta förvärrar effekten av minskad insulinproduktion på leverns glukosproduktion, men är sannolikt ett sekundärt fenomen eftersom det kan korrigeras genom fysiologisk insulinersättning.
δ-cellförändringar och somatostatinproduktion
δ-celler i de Langerhanska öarna frisätter somatostatin, ett hormon som hämmar sekretionen av insulin och glukagon. Forskningen om förändringar i δ-cellernas population och funktion vid T2DM är begränsad. Vissa studier har visat en ökad δ-cellmassa, medan andra inte har noterat några signifikanta förändringar. Studier på djurmodeller har rapporterat ökad somatostatinfrisättning.
Trots dessa observationer är det osannolikt att förändringar i δ-cellmassan eller somatostatinproduktionen är en primär orsak till den minskade insulinfrisättningen och den relativt förhöjda glukagonproduktionen vid T2DM. Snarare kan dessa förändringar vara sekundära och återspegla andra underliggande patofysiologiska processer.


