
Introduktion #
Ur ett historiskt perspektiv har den autoimmuna grunden för typ 1 diabetes (T1DM) främst baserats på tre centrala observationer som tydligt illustrerar sjukdomens patogenes: förekomsten av en inflammatorisk infiltrering i de pankreatiska Langerhanska öarna (så kallad insulit), genetisk predisposition kopplad till det stora histokompatibilitetskomplexet (MHC), samt autoantikroppar riktade mot β-cellantigener.
Dessa koncept utgjorde fundamentet för en modell över T1DM:s naturliga förlopp, som utvecklades under mitten av 1980-talet. Modellen har sedan dess tjänat som en vägledning över generationer för att bättre förstå sjukdomens patogenes, bedöma risken för insjuknande, och utveckla metoder för att förhindra β-cellförlust och bevara endokrin funktion.
Trots att modellen har modifierats över tid, har den bidragit med viktiga framsteg. Dessa inkluderar möjligheten att indela prediabetes i olika stadier (dvs. stadium 1 och 2) och därmed möjliggöra terapeutiska interventioner innan sjukdomen når stadium 3, utveckling av prediktiva biomarkörer, och riktlinjer för studier av miljöfaktorer som påverkar sjukdomsutvecklingen. Modellen har även öppnat upp för en djupare förståelse av sjukdomens mekanismer och dess riskfaktorer.
Trots dessa framsteg återstår dock en betydande terapeutisk utmaning: praktiska svårigheter att förhindra T1DM hos individer med hög sjukdomsrisk eller att långsiktigt bevara β-cellfunktionen hos patienter med nyligen diagnostiserad T1DM.
Hittills är endast en behandling, en anti-CD3-antikropp (teplizumab), godkänd i USA för att bromsa β-cellförlust och försena progressionen till stadium 3 T1DM med cirka två år hos individer med högsta sjukdomsrisk. Ytterligare framsteg på detta område kräver en förbättrad förståelse av sjukdomens patogenes, vilket utgör en avgörande grund för att utveckla effektiva preventiva och terapeutiska strategier.
β-celler #
Under många decennier har T1DM betraktats som en autoimmun sjukdom som främst drivs av autoreaktiva T-celler. Denna uppfattning är fortfarande giltig, men nyare forskning har visat att β-cellerna inte enbart är passiva offer för en autoimmun attack. Flera observationer tyder på att β-cellerna själva spelar en mer aktiv roll i sjukdomsprocessen än vad som tidigare antagits.
Den begränsade långtidseffekten av immunmodulerande behandlingar, förekomsten av inflammatorisk aktivitet i β-cellerna (insulit) utan att individen utvecklar T1DM, samt den stora variationen i insulitgrad vid diagnos och det faktum att en betydande andel β-celler fortfarande finns kvar vid diagnos, har lett till en omvärdering av vår förståelse för T1DM:s patogenes.
Funktion hos β-celler
- Glukosupptag och Metabolism: β-cellerna uttrycker GLUT2, en glukostransportör med låg affinitet men hög kapacitet. Detta möjliggör för cellerna att snabbt reagera på förändringar i blodsockernivåer. När glukos tas upp i cellen fosforyleras det av enzymet glukokinas, vilket är den hastighetsbegränsande reaktionen i glukosmetabolismen. Denna process leder sedan till ATP-produktion via glykolysen.
- ATP-känsliga Kaliumkanaler: En ökad ATP/ADP-kvot leder till att ATP-känsliga kaliumkanaler (K+-ATP-kanaler) stängs. Detta resulterar i en depolarisering av cellmembranet.
- Inflöde av kalcium och Insulinsekretion: Depolariseringen av cellmembranet leder till öppnandet av spänningsberoende kalciumkanaler, vilket ger upphov till ett inflöde av kalciumjoner. Det förhöjda intracellulära kalciumutbudet utlöser sedan exocytos av insulininnehållande granuler, vilket resulterar i frisättning av insulin.
Hormoner som utsöndras av friska β-celler
- Amylin: Amylin utsöndras samtidigt som insulin och har flera viktiga funktioner. Det saktar ner magsäckstömningen, hämmar frisättningen av glukagon och bidrar till en ökad mättnadskänsla.
- Insulin: Insulin är det primära anabola hormonet och spelar en central roll i metabolismen. Det främjar glukosupptag i celler, glykogensyntes i lever och muskler, lipidlagring i fettvävnad och proteinsyntes i muskler.
- C-peptid: C-peptid utsöndras i ekvimolära mängder tillsammans med insulin och används som en markör för β-cellfunktion. Det är särskilt användbart för att bedöma den kvarvarande β-cellkapaciteten hos patienter med T1DM.
Hormonell reglering av β-celler
β-cellerna påverkas av en mängd olika hormoner och parakrina signaler:
- Glukagon: Utsöndras av alfaceller i de Langerhanska öarna och ökar blodsockernivåerna genom att stimulera glykogenolys och glukoneogenes. Glukagon modulerar också β-cellfunktionen.
- Somatostatin: Utsöndras av delta-celler och fungerar som en lokal negativ återkoppling genom att hämma frisättningen av både insulin och glukagon.
- Inkretiner (GLP-1, GIP): Dessa hormoner, som utsöndras från tarmen, förstärker den glukosstimulerade insulinsekretionen och främjar β-cellöverlevnad.
- Adrenalin: Via alfa-adrenerga receptorer hämmar adrenalin insulinfrisättning, särskilt under stress eller vid hypoglykemi.
- Kortisol och Tillväxthormon: Dessa hormoner motverkar insulineffekter och bidrar till att spara glukos genom att stimulera glukoneogenes och minska glukosupptag i perifera vävnader.
Anatomi och normalfysiologi av β-cellerna #
β-cellerna är specialiserade endokrina celler som finns i bukspottkörtelns Langerhanska öar. Dessa öar utgör cirka 1–2 % av bukspottkörtelns totala volym och är utspridda i bukspottkörtelvävnaden. En frisk vuxen bukspottkörtel innehåller ungefär en miljon Langerhanska öar, där β-cellerna står för 60–80 % av de endokrina cellerna. Denna organisation möjliggör en effektiv och snabb respons på förändringar i blodsockernivåer och andra metaboliska signaler.
β-cellernas roll i T1DM är mer komplex än vad som tidigare antagits. De är inte enbart passiva offer för en autoimmun attack utan kan också bidra aktivt till sjukdomsprocessen. Deras förmåga att känna av och reagera på förändringar i blodsockernivåer, samt deras interaktion med andra hormoner och signalmolekyler, gör dem till en central del av metabolismens reglering.
En fördjupad förståelse för β-cellernas funktion och deras roll i T1DM är avgörande för att utveckla nya och mer effektiva behandlingsstrategier.
Langerhanska öar och deras celltyper
Langerhanska öar består av flera olika endokrina celltyper, var och en med specifika funktioner:
- Alfaceller: Producerar glukagon och är perifert placerade i öarna. Glukagon motverkar β-cellernas funktion genom att höja blodsockernivåerna.
- Deltaceller: Producerar somatostatin, som reglerar både alfa- och β-cellernas aktivitet.
- PP-celler: Producerar pankreatiskt polypeptid (PP), som modulerar gastrointestinal aktivitet.
- Epsilonceller: Producerar ghrelin och spelar en mindre roll i endokrin reglering.
β-cellernas struktur och funktion
β-cellerna är polygonala till formen och har ett välutvecklat endoplasmatiskt retikulum och Golgiapparat, vilket speglar deras roll i proteinsyntes. De innehåller tätt packade sekretoriska granuler där insulin, deras primära sekretionsprodukt, lagras i inaktiv form (proinsulin) och senare bearbetas till aktivt insulin och C-peptid.
I Langerhanska öarna är β-cellerna ofta centralt placerade och omges av andra endokrina celler, vilket underlättar en snabb och koordinerad respons på metaboliska förändringar.
Betacellsfunktion vid och efter diagnos av T1DM
- Återhämtning av betacellsfunktion: Isolerade öar från pankreasbiopsier i den norska DiViD-studien visade återhämtad funktion i kultur. Detta antyder att vissa β-cellerna kan vara funktionellt dämpade snarare än permanent förstörda, vilket öppnar möjligheter för terapeutiska interventioner.
- Kvarstående betacellsfunktion vid diagnos: Vid klinisk diagnos av T1DM finns det ofta kvarvarande β-cellerna som fortfarande producerar insulin. Studier har visat att upp till 40–60 % av öarna kan färgas positiva för insulin hos äldre barn och vuxna med nyligen diagnostiserad T1DM. Denna kvarvarande funktion bekräftas också av stimulerade C-peptidsvar, vilka kan kvarstå i många år efter diagnos.
- Långsiktig progression av T1DM: T1DM är en kronisk sjukdom där autoimmuniteten fortskrider asynkront över tid. Förstörelsen av β-cellerna är inte komplett vid diagnos utan fortsätter under många år. Låga nivåer av β-cellproliferation och apoptos tyder på viss dynamik i β-cellmassan, även vid långvarig sjukdom.
Typ 1 diabetes: en autoimmun sjukdom #
Ö-cellsautoantikroppar riktar sig mot Langerhanska öar, särskilt autoantigener på de insulinproducerande β-cellerna. Dessa autoantikroppar uppträder före sjukdomsdebut och kan kvarstå i flera år efter diagnos, dock i mycket lägre nivåer.
Därför är autoantikroppar tillförlitliga prediktorer för T1DM, även om en mindre andel individer med antikroppar inte utvecklar sjukdomen. De viktigaste autoantikropparna är GAD, IA-2, IAA och ZnT8, medan andra är mindre specifika eller känsliga och därför inte användbara som markörer.
Miljöfaktorer
Även om sambandet mellan miljöfaktorer och T1DM är indirekt, finns det bevis för att vissa virus och kostfaktorer kan påverka sjukdomsutvecklingen. Kostrelaterade faktorer inkluderar tidig exponering för komjölk, glutenhaltig kost, kort amningsperiod och vissa livsmedelstillsatser.
Virusinfektioner har också kopplats till sjukdomen. Detta baseras på samband mellan säsongsbundna T1DM-fall och virala epidemier samt närvaro av proinflammatoriska cytokiner som IFN-α hos T1DM-patienter före infektion. Starkast stöd finns för rubella, som kan orsaka direkt destruktion av β-celler, men även coxsackievirus och vissa enterovirus har föreslagits som triggers.
Den tidiga immunologiska processen som leder till insulit vid T1DM initieras av det medfödda immunsystemet och involverar aktivering av mönsterigenkänningsreceptorer (PRRs) av endogena signaler eller exogena ligander som produceras under virusinfektioner i β-cellerna.
Detta är en möjlig koppling mellan miljöfaktorer och utvecklingen av T1DM. Aktiviteten leder till produktion av typ I-interferon, såsom IFN-α, i β-celler och andra celler i de Langerhanska öarna, vilket rekryterar immunceller till platsen.
Tidig och senare immunrespons vid insulit
- Makrofager är först på plats och producerar höga nivåer av tumörnekrosfaktorer (TNF), vilket aktiverar NF-κB i β-celler och utlöser apoptos.
- Senare stadier karaktäriseras av en inflammatorisk mikromiljö i pankreasöarna, vilket ökar kärlpermeabiliteten och möjliggör infiltration av både naiva och aktiverade T-celler. Infiltratet domineras av CD8+ T-celler, men inkluderar också CD20+ B-celler, CD4+ T-celler och CD68+ makrofager.
Den inflammatoriska miljön driver β-celler till att aktivera antiinflammatoriska signalvägar, såsom IL-10 och IL-4/13, samt uttryck av immunkontrollproteiner som PD-L1 och HLA-E. Samtidigt påverkar proinflammatoriska cytokiner som IL-1β och IFNγ β-cellernas metabolism, elektriska aktivitet, insulinsyntes och gap junction-kopplingar. Detta leder även till överdriven produktion av reaktiva syreföreningar (ROS) och aktivering av kaspaser, vilket ytterligare förvärrar β-cellsskadorna.
Två sjukdomsendotyper: T1DM-E1 och T1DM-E2
Studier har identifierat två distinkta endotyper av T1DM med åldersrelaterade skillnader:
1. T1DME1: Tidig debut (oftast hos yngre individer), associerad med HLA-DR4/DQ8-alleler och tidigt framträdande av anti-insulin-antikroppar (IAA). Histopatologiska kännetecken inkluderar:
- Kraftig insulit med hög förekomst av CD8+ T-celler och CD20high B-celler.
- Begränsat antal insulininnehållande öar (ICIs).
- Avvikande insulinprocessering i β-celler.
2. T1DME2: Sen debut (över 13 års ålder), oftare kopplad till HLA-DR3/DQ2-alleler och tidigt framträdande av anti-GAD-antikroppar (GADA). Histopatologiska kännetecken:
- Milda insulitdrag med låga nivåer av CD8+ T-celler och CD20low B-celler.
- Fler kvarvarande ICIs och normal insulinprocessering.
- Mindre tydlig roll för T-celler i patogenesen.
β-cellernas roll i sjukdomsprogression
β-celler är inte enbart passiva mål i sjukdomsprocessen utan bidrar aktivt till den autoimmuna attacken:
- Stressrespons och proteinhantering: Vid ökad insulinsyntes aktiverar β-celler den ofoldade proteinresponsen (UPR) i endoplasmatiska retiklet för att återställa homeostas. Dysfunktion i denna process leder till ansamling av felveckade proteiner som presenteras som neoepitoper via HLA-I och triggar autoimmunitet.
- Inflammation och dedifferentiering: Proinflammatoriska cytokiner, såsom IFNα, IL-1β och IFNγ, inducerar dedifferentiering av β-celler, vilket resulterar i minskad insulinsyntes och förlust av β-cellsspecifika gener.
- Antigenpresentation: HLA-I och HLA-II uttrycks onormalt högt i β-celler hos personer med T1D, vilket gör att β-celler fungerar som antigenpresenterande celler (APCs). Detta förstärker rekryteringen av autoreaktiva CD8+ T-celler.
Mekanismer för autoimmun aktivering
- Neoantigener och T-cellsaktivering: Vid stress, såsom virusinfektioner, genereras hybrid- eller kimeriska neoepitoper som bidrar till aktivering av diabetogena CD4+ och CD8+ T-celler.
- Systemisk kommunikation: Stressade β-celler utsöndrar autoantigener som preproinsulin och exosomer innehållande proteiner som GAD56 och IA-2 till blodcirkulationen. Detta underlättar antigenupptag av APCs utanför pankreas och förstärker immunresponsen.
1. Utveckling och rekrytering av immunceller
- Benmärg: Precursorceller för T- och B-lymfocyter härstammar från benmärgen. B-celler migrerar till mjälten och lymfoida vävnader där de mognar och differentierar.
- Thymus: T-cellsprecursorer går in i thymus för utbildning. Under denna process elimineras normalt autoreaktiva T-celler som känner igen kroppsegna antigener, såsom ö-specifika peptider. Hos individer med predisposition för T1DM undgår dock vissa autoreaktiva T-celler denna elimination.
2. Perifer autoimmun respons
- Autoreaktiva T-celler och omogna B-celler migrerar till lymfoida vävnader, inklusive de lymfkörtlar som dränerar bukspottkörteln.
- Dessa celler stöter på ö-specifika antigener, såsom insulin, GAD (glutamatdekarboxylas) och IAPP (islet amyloid polypeptide). Presentationen av dessa antigener leder till:
- Differentiering av autoreaktiva B-celler till plasmaceller som producerar specifika autoantikroppar.
- Aktivering och proliferation av autoreaktiva T-celler.
3. Tarmens och mikrobiotans roll
- Förändrad tarmmikrobiota och ökad tarmpermeabilitet (”läckande tarm”) tillåter främmande antigener, såsom viruspartiklar, att komma in i systemcirkulationen.
- Dessa antigener kan trigga molekylär mimicry eller förstärka autoimmuniteten genom att aktivera antigenpresenterande celler, såsom makrofager, vilka sedan presenterar ö-antigener för T-celler.
4. Antigenpresentation och inflammation i öarna
- I bukspottkörteln uppreglerar stressade β-celler uttrycket av klass I HLA-molekyler. Detta ökar antigenpresentationen och gör det möjligt för ö-specifika T-celler att känna igen och attackera β-celler.
- Stress på β-celler kan orsakas av miljöfaktorer, såsom virusinfektioner eller inflammation, vilket leder till produktion av hybrid-neoantigener (t.ex. modifierade former av insulin eller IAPP) som ytterligare driver immunaktivering.
5. Destruktion av β-celler
- Autoreaktiva CD8+ T-celler infiltrerar de Langerhanska öarna och riktar sig mot insulinproducerande β-celler. Detta skapar en inflammatorisk miljö, kallad insulit.
- Den fortgående destruktionen av β-celler minskar insulinproduktionen och leder gradvis till hyperglykemi och klinisk diabetes.
6. Autoantikroppar
- B-celler och plasmaceller producerar autoantikroppar mot ö-specifika antigener, såsom insulin, GAD och IAPP. Dessa autoantikroppar är markörer för autoimmunitet men kan också bidra till att förstärka den immunmedierade skadan.
7. Genetiska och miljömässiga utlösare
- Genetisk predisposition, särskilt kopplad till HLA-klass II-lokus, spelar en betydande roll för den selektiva presentationen av ö-antigener till autoreaktiva T-celler.
- Miljöfaktorer, såsom virusinfektioner och tarmmikrobiotans obalans, fungerar som utlösare som förvärrar autoimmuniteten eller stör den immunologiska toleransen.
8. Progression till klinisk diabetes
- Destruktionen av β-celler når en kritisk tröskel, vilket leder till en otillräcklig mängd insulin för att reglera blodsockernivåerna.
- Vid diagnos är en betydande del av β-cellsmassan redan förlorad, även om vissa dysfunktionella β-celler kan kvarstå.
β-cellsdestruktion #
Sjukdomsprogression från ö-cellsreaktion till symtom
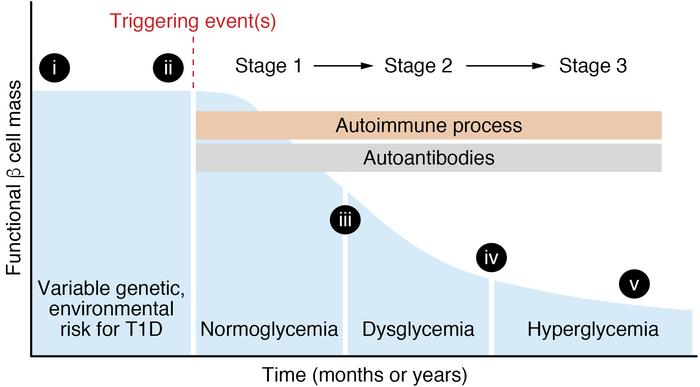
Figuren ovan visar hur hyperglykemi utvecklas hos individer med T1DM när endast cirka 10–15 % av β-cellsmassan kvarstår. Dessa celler är ansvariga för insulinproduktion och frisättning. Sjukdomsprogressionen till avancerat stadium (stadium 3) påverkas av en kombination av miljöfaktorer, genetiska faktorer och förekomst av autoantikroppar riktade mot β-cellerna.
Det är fortfarande oklart vilka faktorer som triggar den autoimmuna reaktionen vid T1DM. Hos cirka 5–10 % av alla nydiagnostiserade patienter saknas autoantikroppar, trots att ö-inflammation (insulitis) kan observeras.
Den autoimmuna processen och insulit
1. Insulitis och immuncellinfiltration: Insulitis är ett patologiskt kännetecken för T1DM, kännetecknat av en inflammatorisk lesion i Langerhans öar med infiltration av immunceller, huvudsakligen lymfocyter. CD8+ cytotoxiska T-celler dominerar och angriper β-celler med hög uttrycksnivå av HLA klass I-antigen. Överuttryck av HLA klass I och II har kopplats till potentiella virusinfektioner som kan bidra till sjukdomsutvecklingen.
2. Heterogenitet i insulitens omfattning: Endast en mindre andel av de Langerhanska öarna (10–30 %) uppvisar insulit, vilket varierar med patientens ålder och tid sedan diagnos. Insulit är vanligare hos yngre patienter och hos dem vars pankreas analyseras nära tidpunkten för diagnos. Vid avancerad sjukdom ses ofta pseudo-atrofiska öar utan insulinproducerande celler.
3. Kronisk inflammation och β-cellsdysfunktion: Insulitiska öar uppvisar ofta ansamling av hyaluronan, en molekyl som underlättar lymfocytadhesion och migration. Dessutom har inflammatoriska mediatorer, såsom cathepsiner, identifierats vid insulitens kant. Dessa ämnen kan underlätta lymfocyternas infiltration i öarna och bidra till β-cellernas destruktion och dysfunktion.
Cellulär autoimmunitet och β-cellsdestruktion #
Genetisk predisposition och antigenpresentation
Den genetiska predispositionen för T1DM innebär en dysreglering av antigenpresentation, en avgörande mekanism för immunsystemet. HLA klass II-molekyler, uttryckta på ytan av antigenpresenterande celler (APC) såsom makrofager och dendritiska celler, binder till T-cellreceptorer (TCR) och aktiverar immunceller.
Vid T1DM leder denna process till en överdriven immunrespons och autoimmun attack på β-celler. Autoreaktiva CD8+ T-lymfocyter är centrala i β-cellernas destruktion, med bidrag från proinflammatoriska cytokiner som ytterligare förstärker skadan.
Insulit och immuncellsinfiltration
Vid obduktion av nyligen diagnosticerade T1DM-patienter har insulit, en inflammatorisk lesion i Langerhanska öar, identifierats. Denna inflammatoriska process omfattar APC, CD4+ och CD8+ T-lymfocyter samt naturliga mördarceller (NK-celler), som alla spelar roller i den autoimmuna processen. Insulitiska öar uppvisar ofta överuttryck av HLA klass I och II, vilket kan vara kopplat till virusinfektioner och deras potentiella roll i sjukdomens patogenes.
Betydelsen av autoantikroppar för sjukdomsprogression
Antalet och typen av ö-cellsantikroppar är viktiga prediktorer för T1DM. Framför allt har autoantikropparna IA-2A och ZnT8A associerats med snabbare progression till klinisk sjukdom. IA-2A är ofta kopplad till högre risk att utveckla flera autoantikroppar och snabbare progression, särskilt hos barn. ZnT8A är också förknippad med utveckling av fler autoantikroppar och högre risk för progression till T1DM.
Däremot har IAA och GADA generellt kopplats till långsammare sjukdomsförlopp. Första gradens släktingar med dessa antikroppar har visat en långsammare progression än de med IA-2A och ZnT8A. Återgång från autoantikroppspositivitet till negativitet är vanligare för GADA och IAA men sällsynt för IA-2A och ZnT8A.
Prediktivt värde av titrar och affiniteter
Höga titrar och affiniteter för IAA och IA-2A har visat sig vara kopplade till snabbare sjukdomsprogression. Studier som DPT-1 och TEDDY har visat att höga nivåer av dessa antikroppar är särskilt prediktiva för T1DM hos barn med genetisk predisposition och autoantikroppspositivitet.
Insulitens roll i sjukdomsutvecklingen
Insulit, karakteriserad av immuncellsinfiltration och inflammatoriska förändringar i β-celler, är en viktig komponent i sjukdomsprocessen. Inflammatoriska mediatorer såsom hyaluronan och cathepsiner bidrar till lymfocyters migration till och destruktion av β-celler. Forskning på pankreasdonatorer med diabetes har ökat förståelsen för insulitens effekter, men vissa fynd kan påverkas av donatorernas kliniska status, såsom stress, ketoacidos och intensivvårdsbehandling.
Cellulär stress och β-cellens respons vid insulit
- Stresstolerans i β-celler: Ökad uttryck av markörer för den ofoldade proteinresponsen (unfolded protein response, UPR) i β-celler under insulit tyder på att adaptiva mekanismer aktiveras för att hantera miljömässig stress. Dessa mekanismer innefattar:
- Minskad total translationshastighet för att begränsa produktionen av nya proteiner.
- Nedbrytning av ackumulerade proteiner i det endoplasmatiska retiklet (ER).
- Ökad produktion av chaperonproteiner som hjälper till att återställa proteinhomeostas.
- Initiering av autofagi för att återställa cellulär jämvikt.
β-cellens sårbarhet
Trots dessa skyddsmekanismer är β-celler särskilt känsliga för inflammatoriska och metabola stressfaktorer på grund av:
- Extrem sekretorisk kapacitet: β-celler kan producera upp till en miljon insulinmolekyler per minut och öka produktionen med över 50 gånger som svar på glukos. Denna höga sekretoriska aktivitet gör β-celler särskilt känsliga för stress på endoplasmatiskt retikulum.
- Låg antioxidativ kapacitet: β-celler har lågt uttryck av superoxiddismutas och andra antioxidativa försvarsmekanismer, vilket gör dem sårbara för reaktiva syreradikaler (ROS).
- Bristande apoptosskydd: Lågt uttryck av anti-apoptotiska faktorer, såsom BCL-2, gör β-celler mer mottagliga för apoptotiska signaler.
Stresseffekter och apoptosmekanismer
Utöver sin cytoprotektiva funktion kan aktivering av ER-stressensorer utlösa en kaskad av händelser som leder till:
- Direkt apoptos: Via aktivering av IRF–STAT1-vägen.
- Nekroptos: Genom aktivering av TNFR1–RIP1-signalering.
- Nekros: Genom ökad produktion av reaktiva syreradikaler.
- Cellulär senescens: β-celler kan utveckla ett senescens-associerat sekretoriskt fenotyp (senescence-associated secretory phenotype, SASP) som bidrar till inflammation.
Molekylär förklaring av stressad β-cell
Vid typ 1-diabetes leder cytokiner till stora förändringar i gen- och proteinuttryck via STAT1-, IRF1- och NF-κB-signalvägar. Dessa orsakar en överuttryckning av HLA klass I och uppreglering av inhibitoriska receptorer som PDL1.
Under stress tas kalcium upp av mitokondrier, vilket ökar deras permeabilitet och frisätter pro-apoptotiska faktorer som reaktiva syreradikaler (ROS) och cyklochrom c. Kalciumförlust i ER (endoplasmatiskt retikulum) aktiverar kalciumberoende enzymer (transglutaminas 2 och peptidylarginindeiminas), vilket medför proteinmodifiering genom deaminering och citrullinering.
ER-stressresponsen aktiverar chaperoner som BiP och sensorer som PERK, IRE1α och ATF6. PERK minskar proteinsyntes genom att fosforylera translationsinitieringsfaktorn eIF2α, medan IRE1α och ATF6 aktiverar nedbrytning av felveckade proteiner och återställer ER-homeostas. Vid långvarig inflammation aktiveras mekanismer som autofagi, selektiv proteinnedbrytning och utsöndring av mikrovesiklar. Detta leder slutligen till apoptos via CHOP, som aktiveras av PERK och ATF6.
Långvarig stress påverkar även transkription, translation och nedbrytning, vilket genererar alternativa RNA-splicingprodukter, defekta ribosomprodukter och hybridpeptider, samlade under termen ”stressed β-cell ligandome”.
Sammanfattning av mekanismer för β-cellsdestruktion
| Mekanism eller Autoantigen | Egenskap/Process |
|---|---|
| Mekanism för β-cell-destruktion | β-celler kan förstöras av miljöfaktorer som virus. Dessa celler fagocyteras sedan av dendritiska celler (antigenpresenterande celler, APC), som aktiveras och migrerar till lymfknutor. Där presenteras antigener till och aktiverar CD4+ och CD8+ T-lymfocyter. CD8+ T-lymfocyter återvänder till blodcirkulationen och bukspottkörtelns öar för att förstöra fler β-celler. Denna process leder till en antigen-spridning (epitope spreading). |
| GAD65 Autoantikroppar | GAD65 är ett enzym som finns i bukspottkörtelns β-celler och producerar GABA, en hämmande signalsubstans. Antikroppar mot GAD65 (GAD65Ab) finns hos 70–80 % av barn med nydebuterad T1DM och är associerade med risk-HLA-typer, såsom DR3-DQ2 och DR4-DQ8. Dessa antikroppar är stabila över tid och kan användas för prediktion och uppföljning av β-cellfunktion. |
| IA-2 Autoantikroppar (IA-2Ab) | IA-2 är ett plasmamembranprotein i bukspottkörtelns β-celler och andra neuroendokrina vävnader. Antikroppar mot IA-2 (IA-2Ab) finns hos 60–70 % av patienter med nydebuterad T1DM och är mindre vanliga hos äldre patienter. Kombinerade tester för IA-2Ab och andra autoantikroppar, såsom GAD65Ab, används för att öka förutsägbarheten av T1DM hos barn. |
| Insulin Autoantikroppar (IAA) | Insulin och dess föregångare proinsulin är β-cell-specifika autoantigener. IAA upptäcks tidigt i sjukdomsförloppet, särskilt hos yngre barn, och är associerade med hög-risk HLA-typer, såsom DR4-DQ8. IAA-nivåer varierar och kan vara lägre hos äldre individer. Tester som radiobindningsanalyser används för att upptäcka IAA, men det finns fortfarande problem med standardisering mellan laboratorier. |
| ZnT8 Autoantikroppar (ZnT8Ab) | ZnT8 är en zinktransportör som finns i β-celler och spelar en viktig roll i kristalliseringen av insulin. ZnT8Ab upptäcks hos 60–80 % av patienter med nydebuterad T1DM och är specifika för T1DM. Dessa antikroppar är särskilt användbara vid prediktion av sjukdomen hos äldre barn. Variationer i ZnT8-genen (SLC30A8) har kopplats till T1DM-risk och påverkar antikroppsproduktionen mot ZnT8. |
| Humoral autoimmunitet | Autoantikroppar mot bukspottkörtelns ö-celler (ICA) identifierades först 1974. Senare utvecklades tester för antikroppar mot specifika autoantigener såsom GAD65, IA-2, insulin och ZnT8. När flera autoantikroppar upptäcks (≥2), särskilt om de är långvariga (>6 månader), ökar risken för T1DM avsevärt. De flesta patienter med T1DM har autoantikroppar flera månader eller år före den kliniska debuten. |
| β-cells autoimmunitet | Kronisk inflammation och stress påverkar β-celler vid typ 1-diabetes genom att aktivera cellulära stressresponser, såsom ER-stress och mitokondriell dysfunktion, vilket leder till proteinmodifiering, försämrad homeostas och slutligen apoptos. Dessa mekanismer försvagar β-cellernas förmåga att hantera stress och bidrar till deras destruktion i sjukdomens patogenes. |
| Triggers | Genetik, virala infektioner (enterovirus, coxsackievirus, cytomegalovirus, rotavirus, EBV), miljöfaktorer (gluten, Vit-D brist, mjölkprotein, nitrater), stress och trauma, toxiner, autoimmun korsreaktion, säsong och geografiska förklaringar. |
Immunmodulerande läkemedel #
Följande läkemedel har studerats för behandling och bromsande and sjukdomsinsjuknande vid T1DM:
- Rituximab: anti-CD20 mAB
- Anti-thymocyte globulin: anti-CD2, CD3 och TCR
- Alefacept: blockerar CD2 kostimulerande receptor
- Abatacept: CTLA-4
- Teplizumab: anti-CD3 mAB (accepterats av FDA för högrisk patienter)
- Golimumab, Etanercept: TNF-blockerare
- Baricitinilib: JAK1/JAK2 hämmare
- Imatinib: Tyrosine kinase hämmare
- IL-21 + liraglutide: IL-21 immunmodulerande effekt
Typ 1 diabetes utan autoimmunreaktion #
T1DM uppstår genom en nästan fullständig förlust av insulin, vilket orsakas av en autoimmun destruktion av de insulinproducerande β-cellerna i bukspottkörteln. Detta leder till kliniska symtom som högt blodsocker.
Hos icke-spansktalande kaukasiska populationer är över 90 % av T1DM av den immunmedierade typen, där HLA-association är dokumenterad och autoantikroppar mot öceller kan upptäckas vid diagnos.
Den återstående 10 %, som ofta benämns idiopatisk diabetes, saknar HLA-association och detekterbara autoantikroppar. Denna typ är vanligare hos andra etniska grupper, såsom asiater, afroamerikaner och latinamerikaner, och tros vara kopplad till virusinfektioner.
Genetik #
Konkordansraten för T1DM bland enäggstvillingar varierar mellan 30–70 % beroende på uppföljningstid, vilket står i kontrast till 10–19 % hos tvåäggstvillingar. Denna skillnad belyser hur både genetiska och miljömässiga faktorer samverkar i sjukdomens utveckling.
Trots att över 85 % av T1DM-fallen inträffar hos individer utan känd familjehistoria, är risken för förstagradssläktingar 15 gånger högre än för den allmänna befolkningen. Risken varierar beroende på vilken förälder som är drabbad:
- En drabbad far: 6–9 % risk för barnet.
- En drabbad mor: 2–4 % risk för barnet.
- Om båda föräldrarna är drabbade: Upp till 30 % risk.
Viktiga genetiska faktorer
Trots att arvsmönstret för T1DM fortfarande inte är helt klarlagt, har omfattande forskning identifierat flera genetiska associationer. Genetiska-studier har bekräftat starka kopplingar mellan T1DM och HLA-systemet (Human Leukocyte Antigen), men minst 47 andra icke-HLA-genetiska faktorer bidrar också till sjukdomsrisken.
Nedan presenteras de mest framträdande genetiska faktorerna som är associerade med T1DM:
Tabell: viktiga genetiska faktorerna associerade med T1DM
| Genetisk faktor | Plats | Beskrivning | Odds Ratio (OR) |
|---|---|---|---|
| HLA-gener | HLA klass II Chr.6p21 | Högsta riskgenotyp: DR3-DQ2/DR4-DQ8 | 18.7 |
| Hög risk Haplotyper | DR4-DQ8 (DRB104-DQA10301-B10302)<br>DR3-DQ2 (DRB103-DQA10501-B10201) | Associerade med ökad risk | 2.0–11.4 2.5–5.0 |
| Skyddande Haplotyper | DR15-DQ6 (DRB115-DQA10102-B10602)<br>DR14-DQ5 (DRB114-DQA10101-B10503) | Associerade med minskad risk | 0.03–0.2 0.02 |
| INS-VNTR | Chr.11p15 | Reglerar central tolerans mot insulin | 2.25 |
| PTPN22 | Chr.1p13 | Protein tyrosinfosfatas non-receptor typ 22 | 1.95 |
| IL2RA (CD25) | Chr.10p15 | Interleukin-2 receptor, alfa-kedjan | 1.70 |
| C12orf30 | Chr.12q24 | Likhet med proteinet KIAA0614 | 1.33 |
| ERBB3 | Chr.12p13 | Erytroblastiskt leukemivirus-onkogen homolog 3 | 1.25 |
| PTPN2 | Chr.18p11 | Protein tyrosinfosfatas non-receptor typ 2 | 1.22 |
| CLEC16A | Chr.16p13 | KIAA0350: C-typ lektindomän familj 16, medlem A | 1.22 |
| CTLA-4 | Chr.2q33 | Cytotoxiskt T-lymfocyt-associerat protein-4 | 1.20 |
| IFH1 (MDA5) | Chr.2q24 | Interferoninducerad med helicas C-domän 1 | 1.15 |
Hög-riskgenotyper och haplotyper
Den genotyp som är starkast kopplad till T1DM är en kombination av två högrisk-haplotyper:
- DR3-DQ2 (DRB103-DQA10501-B1*0201)
- DR4-DQ8 (DRB104-DQA10301-B1*0302).
Dessa haplotyper hittas hos över 95 % av individer med T1DM som diagnostiserats före 30 års ålder, men också hos 40–50 % av den allmänna befolkningen. När dessa haplotyper ärvs tillsammans sker en synergistisk effekt som avsevärt ökar risken för T1DM.
Exempelvis är DQ8 (DQA10301-B10302) oftast associerad med specifika varianter av DRB1*04, såsom:
- DRB1*0401
- DRB1*0404
- DRB1*0402
Däremot har DRB1*0403 en skyddande effekt och är negativt associerad med T1DM. Å andra sidan är DQ2 (DQA10501-B10201) starkast kopplad till DRB1*03.
Vissa alleler, såsom DQB1*0302, DRB1*03 och DRB1*0401, är oberoende riskfaktorer, medan andra alleler och haplotyper kan ge skydd. Exempel på skyddande haplotyper inkluderar:
- DQ6 (DQA10102-B10602 och DQA10102-B10603)
- DQA10101-B10503
- DQA10202-B10303
Dessutom har andra HLA klass II-alleler, såsom DPB1, och vissa klass I-alleler också kopplats till T1DM-risk, och forskning pågår för att identifiera nya associationer.
Andra genetiska faktorer
Utöver HLA-regionen har flera icke-HLA-gener identifierats som bidrar till T1DM-risken, även om deras effekt är mindre än HLA-haplotyperna. Några av de viktigaste generna är:
- INS-VNTR på kromosom 11: reglerar immunologisk tolerans mot insulin.
- PTPN22 (LYP) på kromosom 1: kodar för ett enzym kopplat till immunsignalering.
- IL2RA (CD25) på kromosom 10: kodar för en del av interleukin-2-receptorn, som är viktig för regleringen av T-celler.
Miljöfaktorer
Även om genetiska faktorer spelar en central roll i T1DM, visar flera observationer att miljöfaktorer också bidrar till sjukdomsutvecklingen. Bland dessa faktorer märks:
- Säsongsvariation: Förekomsten av diabetes visar en koppling till årstid och födelsedatum, där fler fall diagnosticeras under vissa årstider.
- Virusinfektioner: Vissa virus har kopplats till ökad risk för T1DM, och infektioner tros kunna utlösa autoimmuna reaktioner hos genetiskt mottagliga individer.
- HLA och utveckling av T1DM: Endast cirka 10 % av individer med genetisk predisposition utvecklar faktiskt sjukdomen, vilket belyser miljöfaktorernas betydelse.
Sammanfattningsvis är T1DM ett resultat av komplexa interaktioner mellan genetiska och miljömässiga faktorer. Förståelsen av hur dessa faktorer samverkar är avgörande för framtida forskning och behandlingar.
Tabell: huvudsakliga miljöfaktorer som ökar risken för T1DM
| Faktor | Föreslagna Effektmekanismer | Exempel |
|---|---|---|
| Maternella faktorer | Triggar autoimmun respons | Graviditetsinfektioner, högre moderns ålder, högre födelseordning, ABO-blodgrupps-inkompatibilitet |
| Okänd mekanism | ||
| Virusinfektioner | Direkt β-cellsskada (cytolys), efterliknande av β-cell-autoantigener (mimikry), aktivering av autoreaktiva T-celler som leder till β-cellförlust | Påverkan på insulinproduktion via uttryck av HLA-gener och interferoner |
| Mässlingvirus, röda hund-virus, enterovirus/Coxsackie B-virus, rotavirus, cytomegalovirus, Epstein-Barr-virus | ||
| Kostfaktorer | Triggar autoimmun respons | Kort amningsperiod/bovint mjölk, spannmål, vitamin D-brist |
| Okänd mekanism | Hög proteinhalt | |
| Faktorer relaterade till insulinkänslighet och/eller insulinresistens | Belastning av β-celler genom ökade krav (”acceleratorhypotesen”) | Pubertet, högkalorikost, viktökning |
| Ökad insulinresistens | ||
| Psykologisk stress | Påverkar hypotalamus-hypofys-binjure-axeln, vilket stör autonoma nervsystemet och leder till autoimmun dysreglering | Stress under graviditet, separation mellan barn och föräldrar, beteendeavvikelser, svår anpassning |
| Toxiska substanser | Direkt skada på β-celler | Alloxan, streptozocin, Vacor |


