
Typ 2 diabetes är en komplex och multifaktoriell sjukdom där både metabola och icke-metabola organ spelar en central roll i sjukdomens utveckling
Typ 2 diabetes mellitus (T2DM) kännetecknas av en dysreglerad metabolism av kolhydrater, lipider och proteiner. Detta tillstånd är en komplex och multifaktoriell sjukdom där både genetiska och miljömässiga faktorer bidrar till dess uppkomst och progression.
Utvecklingen av T2DM kan härledas till en eller flera av följande mekanismer: 1) försämrad insulinsekretion, 2) reducerade insulindepåer, samt 3) insulinresistens, eller en kombination av dessa faktorer.
Vetenskapen kring T2DM är komplex då den präglas av motstridiga resultat från olika studier. Trots detta framstår en progressiv försämring av insulinsekretionen från β-celler i pankreas som en central mekanism bakom sjukdomsutvecklingen.
Denna dysfunktion är sannolikt starkt kopplad till en underliggande insulinresistens som involverar skelettmuskulatur, lever, fettvävnad och pankreas. Ofta föregås denna process av prediabetes, ett hög-risktillstånd som markant ökar risken för att utveckla T2DM.
Patofysiologin omfattar flera komponenter: #
- β-cellfunktion: En progressiv försämring av β-cellernas funktion, med minskad känslighet för glukagonliknande peptid-1 (GLP-1) och reducerad insulinproduktion.
- Leverinsulinresistens: Ökad känslighet för glukagon, vilket resulterar i ökad endogen glukosproduktion och förvärrad hyperglykemi.
- Fettvävnad: Accelererad lipolys leder till förhöjda nivåer av fria fettsyror, vilka bidrar till insulinresistens i muskler och lever samt försämrar β-cellfunktionen ytterligare.
- Njurfunktion: Förhöjd glukosreabsorption via natrium-glukos-kotransportör 2 (SGLT2) i njurarna förstärker hyperglykemin.
- Centrala nervsystemet (CNS): Dysreglerad hormonell aptitkontroll, inklusive nedsatt aptitdämpande effekt av insulin, bidrar till viktuppgång och ökad insulinresistens.
- Inflammation: Kronisk låggradig inflammation, med frisättning av proinflammatoriska cytokiner och interleukiner från fettvävnad, bidrar till systemisk inflammation. Denna inflammatoriska miljö påverkar insulinets signalvägar negativt och förvärrar insulinresistensen i flera metabola vävnader.
- Mitokondriell dysfunktion: Mitokondrier spelar en central roll i cellens energiomsättning, och vid typ 2-diabetes observeras en minskning i mitokondriernas antal, funktion och effektivitet. Detta leder till nedsatt oxidativ fosforylering och ökad produktion av reaktiva syreföreningar (ROS), vilket orsakar cellulär stress, insulinresistens och ytterligare β-cellsdysfunktion.
- Mikrobiomet: Tarmens mikrobiota har visat sig vara en viktig modulator av systemisk metabolism. Vid typ 2-diabetes observeras en förändring i mikrobiell sammansättning, med minskad diversitet och ökad förekomst av patogena bakteriestammar. Detta bidrar till ökad permeabilitet i tarmbarriären, vilket leder till att inflammatoriska molekyler som lipopolysackarider (LPS) läcker ut i cirkulationen och främjar insulinresistens och inflammation.
Insulinresistens: orsaker och konsekvenser #
Figuren nedan visar insulins signalväg hos friska
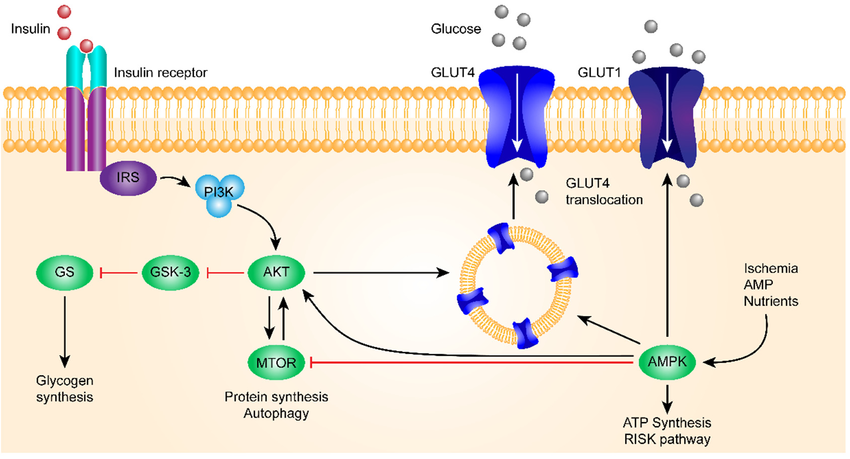
Borros Arneth et al. Int. J. Mol. Sci 2019, 20, 2467; doi:10.3390/ijms20102467
1. Insulin binder till insulinreceptorn (IR) som består av två extracellulära α-subenheter som ansvarar för insulinbindning, dessutom består IR av två membranbundna β-subenheter med intracellulära domäner som innehåller tyrosin-kinasaktivitet. Ett adaptorprotein (insulinreceptorsubstrat-1, IRS-1) fosforylerar tyrosin-kinas på IR som skapar bindningsplatser för signaleringsmolekylen PI3K.
2. PI3K (fosfatidylinositol-3-kinas) är en heterodimer bestående av en katalytisk subenhet (p110) och en regulatorisk subenhet (p85). Den senare medierar bindningen till IRS-1.
3. AKT (även kallad PKB, proteinkinas B) är ett serin/treoninkinas som aktiveras av PIP3. Det reglerar flera nedströms målproteiner, inklusive GSK-3β.
4. GSK-3β (glykogensyntaskinas-3β) är ett viktigt enzym som reglerar glykogensyntes genom att hämma glykogensyntas när det är aktivt. Insulin hämmar GSK-3β via AKT, vilket möjliggör glykogensyntes.
5. AKT och aktivering av GLUT4: AKT fosforylerar AS160, en Rab-GTPas-aktiverande protein (Rab-GAP). Fosforylering av AS160 inaktiverar dess GAP-aktivitet, vilket gör att specifika Rab-proteiner förblir i deras aktiva, GTP-bundna form. Aktiva Rab-proteiner främjar vesikeltransport och fusion av GLUT4-vesiklar med plasmamembranet.
6. Translokation av GLUT4: GLUT4, som normalt finns lagrat i intracellulära vesiklar (GLUT4-lagringsvesiklar, GSV), transporteras till plasmamembranet via vesikulär transport. Vesiklarna fuserar med plasmamembranet, vilket exponerar GLUT4 och möjliggör glukosupptag från blodet.
Insulinresistens vid fetma och typ 2 diabetes mellitus
1. PI3K-vägen och serinfosforylering av IRS-proteiner
- Insulinresistensmekanism: Ökad serinfosforylering av insulinreceptorsubstrat (IRS) blockerar tyrosinfosforylering, vilket hämmar insulinsignaleringen. Detta förstärks ytterligare genom ökad IRS-nedbrytning.
- Orsaker: Ektopisk lipidackumulering, mitokondriell dysfunktion, inflammation och ER-stress är centrala bidragande faktorer.
2. Lipidackumulering och PKC-aktivering
- Ektopiska lipider: Ackumulation av diacylglycerol (DAG) i muskler och lever aktiverar protein kinase C (PKCθ i muskler, PKCδ och PKCε i levern), vilket fosforylerar IRS-proteiner och hämmar insulinsignaleringen.
3. Mitokondriell dysfunktion
- Effekter: Minskad mitokondrietäthet och störd oxidativ fosforylering bidrar till ökad produktion av reaktiva syreradikaler (ROS), som aktiverar serinkinaser och inducerar insulinresistens.
- Adipokiner: Mitokondriell dysfunktion i fettväv minskar adiponektinutsöndring, vilket förvärrar insulinresistens.
- Om mitokondriell dysfunktion är en orsak eller konsekvens av insulinresistens är fortfarande oklart.
4. Inflammation och immunrespons
- Systemisk inflammation: Proinflammatoriska cytokiner (t.ex. IL-1, IL-6 och TNF) och infiltrering av M1-makrofager i fettväv och lever aktiverar signalvägar (IKKβ, JNK1) som hämmar IRS-proteiner. Blockering av TNF förbättrar insulinresistens i djurmodeller, men effekten hos patienter är begränsad.
- Makrofager och T-celler: Obalans mellan proinflammatoriska M1-makrofager och antiinflammatoriska M2-makrofager bidrar till lipolys, glukoneogenes och systemisk insulinresistens.
5. Endoplasmatiskt retikulum (ER)-stress och unfolded protein response (UPR)
- ER-stress: Obalans mellan proteinsyntes och bearbetning i ER aktiverar UPR via sensorer som IRE1α, PERK och ATF6α. Detta leder till JNK-aktivering, som bidrar till insulinresistens.
- Orsaker: Fettsyror och hyperlipidemi driver ER-stress och UPR-aktivering. Viktminskning och kemiska chaperoner minskar ER-stress och förbättrar insulinsensitivitet.
6. mTOR-signalering
- mTORC1 och IRS-nedbrytning: Överaktivering av mTORC1 undertrycker insulinreceptor-signalering genom att hämma tyrosinfosforylering och påskynda nedbrytning av IRS1.
- XBP1 och ER-stress: PI3K interagerar med XBP1 och initierar transkription av gener som förvärrar ER-stress och insulinresistens.
Insulinresistens i skelettmuskulatur
Forskning har visat att nedsatt insulinstimulerad glykogensyntes i skelettmuskulaturen är en central faktor bakom insulinresistens. Denna defekt kan spåras till störningar i insulinsignalering på nivåerna av insulinreceptorn (IR) och insulinreceptorsubstrat-1 (IRS-1).
Individer med T2DM uppvisar även reducerad mitokondriedensitet och funktion i muskelceller, vilket förhindrar effektiv lipidoxidation och bidrar till ackumulering av lipider, såsom diacylglycerol (DAG) och triacylglycerol (TAG).
Icke-alkoholorsakad fettlever (NAFLD) och insulinresistens
Ökade nivåer av leverceramider och DAG har visat sig korrelera med oxidativ stress, inflammation och insulinresistens. Nya studier pekar på att dessa effekter delvis beror på nedsatt mitokondriefunktion och förändringar i lipidsignalering, inklusive aktivering av protein kinas C (PKC) och fosfoinositid-3-kinas (PI3K). Samtidigt är NEFA-flödet till levern en viktig drivkraft bakom NAFLD och associerade metabola störningar.
Hyperinsulinemi och kompensationsmekanismer
En viktig aspekt av insulinresistens är förekomsten av hyperinsulinemi, där blodinsulinnivåerna är högre än normalt i förhållande till glukosnivåerna, både i fasta och efter måltid. Hyperinsulinemi är en kompensationsmekanism för insulinresistens i perifera vävnader och syftar till att normalisera blodglukosnivåerna.
När bukspottkörteln misslyckas med att producera tillräckligt mycket insulin för att möta detta ökade behov uppstår en betydande störning i kroppens glukoshomeostas. Detta resulterar i hyperglykemi och glukosintolerans, som utmärker T2DM.
Intressant nog bibehåller många personer med T2DM en relativ hyperinsulinemi fram till de sena stadierna av sjukdomen. På molekylär nivå karakteriseras insulinresistens av en nedsatt förmåga hos insulin att aktivera glukostransport i muskel- och fettceller på grund av en dysfunktion i GLUT4-glukostransportsystemet i dessa vävnader.
Dessutom är ett kännetecken för insulinresistens att leverns glukosproduktion inte kan hämmas effektivt, vilket till stor del beror på en ihållande ökning av glukoneogenesen, potentiellt som en indirekt följd av dysreglerad lipidmetabolism i fettvävnad.
Övergången från normoglykemi till hyperglykemi
Långtidsstudier har visat att personer som senare utvecklar typ 2-diabetes (T2D) uppvisar en gradvis ökning av både faste- och postprandiella glukosnivåer. Insulinkänsligheten, som påverkas av ålder, kön och viktuppgång, börjar minska flera decennier innan sjukdomsdebut och representerar en av de tidigaste patogenetiska förändringarna.
Denna minskning beror främst på försämrad icke-oxidativ glukosmetabolism, särskilt nedsatt insulinstimulerad inlagring av kolhydrater som muskelglykogen. För att kompensera för insulinresistensen ökar β-cellerna sin insulinutsöndring, vilket leder till hyperinsulinemi. Detta främjar leverns de novo-lipogenes (DNL), steatos, hyperlipidemi och tillväxt av vit fettvävnad (white adipose tissue, WAT).
WAT-dysfunktion, som orsakas av insulinresistens och inflammation, ökar lipolysen och förvärrar därmed leverns insulinresistens och icke-alkoholorsakad fettlever (NAFLD). Dessutom stimulerar ökat flöde av fria fettsyror (NEFA) och glycerol till levern glukoneogenesen. Kombinationen av avtagande β-cellsfunktion, delvis orsakad av glukolipotoxicitet, och ökade glukosnivåer leder slutligen till faste- och postprandiell hyperglykemi.
Postprandiella levermetaboliter och insulinresistens
Fastehyperglykemi vid T2D beror på ökad hepatisk glukoneogenes och endogen glukosproduktion (EGP), främst på grund av insulinresistens i levern. Detta karakteriseras av insulins nedsatta förmåga att hämma dessa processer. Ny forskning visar att dessa indirekta effekter sannolikt är medierade via insulinets verkan på WAT, vilket förklarar den akuta suppressionen av glukoneogenesen under hyperinsulinemi efter måltid.
Studier med 13C-magnetresonansspektroskopi har visat att personer med T2D har nedsatt postprandiell och insulinreglerad leverglykogensyntes. Nedsatt aktivering av insulinreceptorn och efterföljande modifieringar i glykogensyntesmaskineriet indikerar en gemensam defekt i insulinets verkningsmekanism. Parallellt är leverns insulinresistens ofta associerad med ökad hepatisk de novo-lipogenes och NAFLD
Insulinresistens utan T2DM
Även utan utveckling av T2DM är individer med insulinresistens predisponerade för allvarliga sjukdomar associerade med diabetes, såsom retinopati, neuropati och njursjukdom. Detta understryker vikten av att utveckla metoder för att tidigt identifiera och hantera insulinresistens för att minska risken för dessa komplikationer.
Forskning visar även att det finns individer med uttalad fetma som inte lider av insulinresistens, dessa kallas för metabolic healthy obese individuals och utgör en liten andel av alla individer med fetma.
Sviktande β-celler #
Figuren nedan visar hur β-celler reagerar på glukos och frisätter insulin hos friska individer
β-cellfunktion och dysfunktion i T2DM
- Normala β-celler: Utgör cirka 60% av cellerna i de Langerhanska öarna och kommunicerar med α-celler och andra celltyper via gap junctions och hormonella signaler. β-celler kan anpassa insulinsekretionen snabbt för att bibehålla glukoshomeostas.
- β-celler vid T2DM: β-cellmassan är reducerad med 30–40% vid obduktion av patienter med T2DM. Förlusten sker via apoptos och dysreglerad autofagi, medan nybildning (neogenes) är osäker. Insulinsekretionen är nedsatt relativt till glukosnivåerna, och β-celler visar försämrad glukossensitivitet samt högre tröskel för insulinutsöndring.
Störd insulinsekretion
Även om fetma ofta leder till insulinresistens är det endast en mindre andel av obesa, insulinresistenta individer som utvecklar T2DM. I både djurmodeller och hos människor är den utlösande faktorn betacellsvikt, som innefattar en minskning av betacellsmassa och en försämring av centrala betacellsfunktioner, såsom glukosstimulerad insulinsekretion (GSIS).
Hos alla däggdjur, inklusive människor, regleras postprandial insulinsekretion på ett bifasiskt sätt genom närings- och hormonsignaler, där glukos är den primära regulatorn. Andra sekretagoger, såsom fria fettsyror, aminosyror eller inkretinregulatorn glukagonlik peptid-1 (GLP-1), fungerar som förstärkare men kräver en tröskelnivå av glukos i blodomloppet för att aktiveras.
Insulinsekretion från pankreatiska betaceller stimuleras av glukosmetabolism, vilket resulterar i en ökning av ATP:ADP-kvoten. Denna ökning leder till stängning av ATP-känsliga kaliumkanaler (KATP), depolarisation av plasmamembranet, aktivering av spänningsstyrda kalciumkanaler och kalciummedierad stimulering av granulexocytos.
KATP-kanalberoende mekanismen, även kallad “triggersignalen”, är särskilt viktig för den första, akuta fasen av insulinfrisättning, som sker under de första tio minuterna efter glukosstimulering. Under den andra, mer långvariga fasen av insulinsekretion har ATP och kalcium mer begränsade roller, vilket tillåter andra glukosderiverade sekundära budbärare, de så kallade “förstärkningssignalerna”, att dominera.
Mitokondriell glukosmetabolism genererar ytterligare signaler utöver förändringar i ATP:ADP-kvoten, vilka är avgörande för att reglera insulinfrisättning. För att förstå betacellernas funktionella svikt vid diabetes krävs kunskap om alla glukosderiverade signaler, både triggersignaler och förstärkningssignaler.
Genetisk känslighet för β-celler
Studier på Zucker diabetiska fettråttor (ZDF-råttor) visar att betacellmassan först ökar dramatiskt som en kompensatorisk mekanism vid fetma. När kroppsvikten fortsätter att öka och insulinresistensen förvärras minskar dock betacellmassan och dess funktion gradvis.
Minskningen av betacellmassan har tillskrivits ökad apoptos, medan flera teorier har föreslagits för den underliggande orsaken till funktionsförlusten (GSIS).
Liknande mekanismer för betacellsvikt har observerats hos människor med diabetes. Hos feta, icke-diabetiska individer är betacellmassan större jämfört med magra kontrollpersoner. Däremot är betacellmassan minskad hos feta patienter med nedsatt fasteglukos eller typ 2 diabetes, och betacellapoptos är mer uttalad hos individer med glukosintolerans eller diabetes.
Genetisk bakgrund spelar en viktig roll i att avgöra betacellernas känslighet för dekompensation och progression till T2DM. Detta har tydligt visats i gnagarmodeller. Till exempel uppvisar BTBR/leptin-ob-möss en defekt proliferation av öar som svar på fetma, vilket leder till svår diabetes, medan C57BL6/leptin-ob-möss kan expandera sin betacellmassa och därmed skyddas mot hyperglykemi.
Metabol överbelastning i β-celler
Långvarig exponering av pankreatiska öar för förhöjda näringsnivåer leder till betacellsdysfunktion och, i förlängningen, betacellsdöd. När isolerade råttöar utsätts för hyperglykemi under flera dagar ökar den basala insulinsekretionen, men förmågan att frisätta insulin som svar på stimulerande glukosnivåer försvinner.
På liknande sätt påverkar höga nivåer av fettsyror inte glukosstimulerad insulinsekretion (GSIS) såvida glukoskoncentrationen inte ligger på eller över en tröskelnivå (vanligtvis runt 8 mM). Dessa fynd har lett till konceptet att betacellsdysfunktion uppstår som en konsekvens av “glukolipotoxicitet” snarare än enbart exponering för glukos eller fettsyror individuellt.
I denna modell ökar glukos malonyl-CoA-nivåerna, vilket hämmar CPT1 (karnitinpalmitoyltransferas 1) och fettsyrans oxidation. Lipidmetaboliter styrs istället mot cytosoliska produkter, såsom ceramider eller förestrade lipider, vilket påminner om de mekanismer som ligger bakom insulinresistens. Dock har senare studier visat att de negativa effekterna av fettsyror på betacellsfunktionen snarare kan bero på ökad fettsyraoxidation än minskad.
Effekter av långvarig fettsyraexponering hos β-celler
Långvarig exponering av råttöar för höga nivåer av fettsyror har rapporterats minska aktiviteten hos pyruvatdehydrogenas (PDH) och glukosoxidation, vilket föreslagits hämma den glukosinducerade ökningen av ATP:ADP-kvoten och därmed försämra GSIS. Nyare studier har dock utmanat detta synsätt.
Långvarig behandling av INS1-celler med oleat orsakade endast en måttlig minskning av glukosoxidation, medan glykolytiskt flöde, citratnivåer och PDH-aktivitet förblev opåverkade. I cellinjen INS1-832/13 och i råttöar påverkades glukosoxidationen inte alls av långvarig behandling med fettsyror. Däremot inducerades β-oxidativa enzymer och fettsyraoxidationen stimulerades.
Lipider och mitokondriell dysfunktion
- Lipider och UCP2:
- Fettsyror ökar uttrycket av uncoupling protein-2 (UCP2), vilket kan leda till läckage av protoner i mitokondrierna, nedsatt ATP-produktion och därmed försämrad insulinsekretion.
- Gluko-lipotoxicitet:
- Kronisk exponering för både glukos och fettsyror påverkar β-cellens funktion negativt genom förändrade metabola flöden.
Mekanism för lipidinducerad dysfunktion
Den framträdande mekanismen bakom lipidinducerad försämring av GSIS involverar aktivering av PC genom dess allosteriska aktivator acetyl-CoA. Acetyl-CoA-nivåerna stiger som en följd av uppreglerad fettsyraoxidation i lipidexponerade celler. Ytterligare bevis för pyruvats roll i lipidinducerad betacellsdysfunktion kommer från studier där en membranpermeabel ester av malat, dimetylmalat (DMM), användes.
Tillsats av DMM till betacellinjer och råttöar förbättrade GSIS och ökade pyruvataktiviteten. När DMM inkluderades i insulinsekretionsanalyser av glukosinsensitiva öar från ZDF-råttor eller lipidpåverkade INS1-832/13-celler noterades en markant förbättring av GSIS i båda fallen.
β-cellens apoptos
- ER-stress:
- Överbelastning av β-cellens proteinsynteskapacitet leder till aktivering av unfolded protein response (UPR) och PERK-vägen, vilket vid långvarig stress kan orsaka β-cellens död.
- Högfettdiet:
- Heterozygota möss med mutationer i UPR-signalering uppvisar ER-förändringar, minskad insulinproduktion och utvecklar diabetes vid högfettdiet.
Amyloidfibriller
- Amylin och amyloidbildning:
- IAPP (islet amyloid polypeptide), ett protein som bildar amyloidfibriller, ackumuleras i pankreasöar vid typ 2-diabetes. Detta bidrar till β-cellens apoptos och funktionella försämring.
- Patologisk modell:
- Ackumulerade amyloidfibriller kan skapa en “perfekt storm” genom att kombinera metabolisk stress och dysfunktionell glukossignalering.
Cerebral reglering av levermetabolism #
Hjärnans höga energibehov och begränsade energilagringskapacitet innebär att dess energiförsörjning via glukos och ketoner är helt beroende av levern och, i viss mån, njurarna under svält, med minimal direkt endokrin reglering. Samtidigt kan hjärnans insulinverkan påverka aptitreglering, humör, kognitiv funktion och möjligen perifer glukosmetabolism.
Hos möss verkar insulin och leptin direkt på hypothalamus för att aktivera proopiomelanokortin (POMC) och hämma Agouti-relaterade proteiner, vilket stimulerar mättnad och energiförbrukning. Hypothalamisk inflammation, som ses vid högre nivåer av glios i mediobasala hypothalamus hos överviktiga gnagare och människor, har föreslagits orsaka central insulin- och leptinresistens, vilket bidrar till överdrivet födointag och viktuppgång.
Hos gnagare har central insulinverkan visat sig minska leverns glukosproduktion, WAT-lipolys och glukagonsekretion samtidigt som muskelglukosupptaget ökar. Dock har kontrollerade studier inte kunnat bekräfta liknande hjärninsulinreglering av leverglukosflöden hos vakna hundar eller människor.
Intranasal insulinadministration hos människor minskade inte fasteglukosproduktion men reducerade något leverfett och ökade ATP-innehåll hos glukostoleranta individer, dock inte hos personer med T2DM. Detta tyder på att hjärnans indirekta roll i interorgan-kommunikation snarare är medierad av metaboliter, adipokiner (t.ex. leptin) och enteroendokrina kretsar (t.ex. GLP-1, ghrelin och FGF-19). Ett exempel är hypoleptinemi-inducerad stimulering av hypotalamus-hypofys-binjureaxeln, vilket ökar WAT-lipolys och därmed leverglukoneogenes under svält.
Systemisk inflammation #
Interleukin-1 (IL-1)
Interleukin-1-familjen, särskilt IL-1 och dess antagonist IL-1ra, spelar en avgörande roll i de inflammatoriska processer som driver insulinresistens och β-cellsdysfunktion vid diabetes. Kliniska och experimentella studier tyder på att IL-1ra har potential som en terapeutisk intervention för att förbättra glykemisk kontroll, bevara β-cellfunktion och minska inflammation.
IL-1 är en av de viktigaste proinflammatoriska cytokinerna och består av två subtyper, IL-1α och IL-1β. Dessa aktiverar IL-1 typ I-receptorn och signalvägar såsom NFκB (nuclear factor kappa-B), IKKβ (inhibitor of NFκB kinase subunit β), samt mitogenaktiverade protein kinaser (ERKs, JNKs och P38).
IL-1 samverkar med andra cytokiner, såsom TNF, för att främja inflammatoriska processer genom att dela intracellulära signalvägar och förstärka varandras produktion.
- IL-1 och insulinresistens:
- IL-1β är förhöjt hos individer med metaboliskt syndrom och hos icke-diabetiska individer med en förhöjd risk för typ 2-diabetes.
- IL-1α och IL-1β kan inducera insulinresistens genom att påverka IRS-1-signalering och reducera GLUT-4-translokation till plasmamembranet.
IL-1 receptorantagonist (IL-1ra)
IL-1ra binder till IL-1-receptorer utan att aktivera dem och fungerar därmed som en antagonist till IL-1:s biologiska effekter. IL-1ra produceras av immun-, lever- och fettvävnadsceller och reglerar inflammatoriska processer via en balans mellan IL-1 och IL-1ra.
- IL-1ra och metabolism:
- Förhöjda nivåer av IL-1ra är korrelerade med insulinresistens och är högre hos patienter med metaboliskt syndrom.
- IL-1ra kan påverka leptinresistens genom att hämma leptins aptitdämpande effekter i hypotalamus.
- Djurmodeller har visat att IL-1ra kan minska fettmassa och öka insulinkänsligheten, trots en minskad insulinsekretion.
IL-1 och β-cellers funktion och överlevnad
- Cytokinernas roll vid β-cellsdysfunktion:
- Vid typ 1-diabetes orsakas β-cellsdestruktion av autoimmuna reaktioner, medan typ 2-diabetes associeras med lokal cytokinproduktion och makrofaginfiltration.
- IL-1β har en central roll i att inducera β-cellapoptos och hämma insulinsekretion. Höga glukosnivåer stimulerar IL-1β-produktion i β-celler, vilket leder till försämrad funktion och celldöd.
- IL-1ra som terapeutiskt mål:
- IL-1ra kan skydda β-celler genom att hämma IL-1β:s inflammatoriska effekter, främja cellöverlevnad och stimulera insulinsekretion.
- Kliniska studier har visat att behandling med rekombinant IL-1ra (anakinra) förbättrar glykemisk kontroll och β-cellfunktion samt reducerar systemisk inflammation.
Interleukin-6:s roll vid insulinresistens och diabetes
Interleukin-6 (IL-6) tillhör gp130-familjen av cytokinreceptorer. Medlemmar i denna familj, såsom IL-11 och leukemia inhibitory factor, delar signalproteinet gp130 i sina receptor-komplex. IL-6 signalering kräver bindning till IL-6-receptorn (IL-6R) och efterföljande homodimerisering av gp130. IL-6R finns i två former: en membranbunden 80 kDa-proteinform och en löslig form (sIL-6R), som möjliggör IL-6-signalering även i celler som saknar transmembranbunden IL-6R.
IL-6 aktiverar flera signalvägar, däribland Janus kinase (JAK)–signaltransduktor och aktivator av transkriptionsvägar (STAT) samt MAPK-vägarna. Denna cytokin produceras av flera celltyper, inklusive fibroblaster, endotelceller, monocyter, makrofager, myocyter och adipocyter. Studier har pekat på att IL-6 kan vara involverad i både fysiologi och patologi relaterad till fetma och insulinresistens vid T2DM.
IL-6 och dess relation till fettvävnad
- Cirka 15–30 % av cirkulerande IL-6 härstammar från fettvävnad i frånvaro av träning eller akut inflammation.
- Sekretionen av IL-6 är högre i visceralt fett än i subkutan fettvävnad och sker främst från den stromavaskulära fraktionen, som består av endotelceller och makrofager.
- IL-6-nivåerna är förhöjda i subkutan fettvävnad hos överviktiga individer och minskar vid viktnedgång.
Kontroverser:
- IL-6:s roll vid fetma är fortfarande omstridd. IL-6-defekta möss har rapporterats utveckla fetma och insulinresistens, vilket tyder på att brist på IL-6 kan bidra till metabola störningar.
- Dock har andra forskargrupper inte kunnat reproducera detta fynd, vilket kan bero på skillnader i djurens genetik, miljö eller diet.
IL-6:s effekter på insulinresponsiva vävnader
Lever
- IL-6 försämrar insulinsignalering genom att hämma tyrosinfosforylering av IRS-1 och IRS-2, vilket resulterar i minskad glykogensyntes.
- Kronisk IL-6-exponering undertrycker även autofosforylering av insulinreceptorn, vilket försämrar insulinkänsligheten i levern.
- Neutralisering av IL-6 med antikroppar har visat förbättrad insulinkänslighet i levern i djurmodeller av insulinresistens.
Fettvävnad
- Akut IL-6-exponering stimulerar glukostransport i adipocyter, medan kronisk exponering inducerar insulinresistens.
- Långvarig behandling med IL-6 minskar uttrycket av IRS-1, GLUT-4 och PPARγ samt insulinstimulerad glukostransport och lipogenes.
- SOCS-3 (suppression of cytokine signaling-3) och mTOR (mammalian target of rapamycin) tros spela centrala roller i dessa effekter genom att hämma insulinsignalering via serinfosforylering av IRS.
Skelettmuskel
- IL-6:s effekter på muskler skiljer sig från dess påverkan på lever och fettvävnad. Fler studier behövs för att fastställa de exakta mekanismerna och betydelsen av IL-6 i muskelvävnad.
Andra cytokiners roll vid metabola störningar
Introduktion till IL-8 och IL-18
Utöver de väletablerade cytokinerna som IL-1, IL-1ra och IL-6, produceras även andra proinflammatoriska cytokiner av fettvävnad, såsom IL-8 och IL-18. Även om dessa cytokiner har studerats mindre omfattande, har forskning visat att deras nivåer ökar vid fetma och att de kan vara involverade i metabola sjukdomar som är kopplade till fetma och typ 2-diabetes mellitus (T2DM).
IL-18: metabol betydelse och paradoxala effekter
- Förhöjda nivåer vid fetma och T2DM:
Cirkulerande nivåer av IL-18 är ökade vid fetma, insulinresistens och T2DM. - Effekter i djurmodeller:
Möss som saknar IL-18 eller dess receptor har visat sig ha ökad kroppsvikt och högre incidens av metabola störningar såsom insulinresistens, hyperglykemi, dyslipidemi och ateroskleros, jämfört med vildtypmöss. Detta antyder att IL-18 spelar en skyddande roll mot metabola rubbningar. - Intracerebral påverkan:
Injicering av rekombinant IL-18 i hjärnan hos transgena IL-18-defekta möss minskar födointaget och glykemin. Dessa effekter medieras sannolikt via STAT3-aktivering, vilket speglar de mekanismer som observerats i IL-6-defekta möss. - Cytokinresistens:
Trots ökade plasmavärden av IL-18 vid fetma och T2DM har det föreslagits att denna ökning är kopplad till en resistens mot cytokinet, vilket försvårar dess skyddande funktion.
Sammanfattning av interleukiner, insulin resistens och typ 2 diabetes
- Fetma leder till ett överskott av fettsyror i vävnader som muskler och lever.
- Dessa överskottsfettsyror omvandlas till acyl-CoA, som vidare metaboliseras till diacylglyceroler och ceramider.
- Diacylglyceroler och ceramider, tillsammans med TNF och interleukiner, bidrar till utvecklingen av insulinresistens.
- Insulinresistens uppstår när IRS fosforyleras vid serinrester, vilket förhindrar dess aktivering via tyrosinfosforylering.
- Detta hämmar aktiviteten hos PI3K, vilket leder till minskat glukosupptag och glykogensyntes i musklerna samt ökad glukosproduktion i levern.
- IL-1α och IL-1β är nyckelinterleukiner som bidrar till insulinresistens genom att försämra insulinsignalering, förändra fosforylering av IRS och minska uttrycket av komponenter involverade i glukostransport.
- IL-1ra (en antagonist till IL-1) föreslås påverka inflammatoriska processer och potentiellt modulera insulinkänslighet.
- IL-6 är en annan viktig interleukin associerad med insulinresistens, särskilt i lever och fettvävnad.
- IL-6 försämrar insulinsignalering i levern genom att hämma tyrosinfosforylering av IRS-1 och IRS-2, vilket minskar glykogensyntesen.
- Kronisk exponering för IL-6 hämmar autophosphorylering av insulinreceptorn och tyrosinfosforylering av IRS-1 och IRS-2.
- I fettvävnad stimulerar akut IL-6-exponering glukostransport, men kronisk exponering inducerar insulinresistens.
- Långvarig behandling med IL-6 minskar uttrycket av IRS-1, GLUT-4 och PPARγ, samt insulinstimulerad glukostransport och lipogenes.
- IL-6 påverkar också lipidmetabolism genom att stimulera lipolys i adipocyter, vilket indirekt kan bidra till insulinresistens.
- IL-8 och IL-18 är ytterligare proinflammatoriska cytokiner som produceras i fettvävnad. IL-18 uppvisar en potentiellt paradoxal roll som både en skyddande faktor och en bidragande faktor till insulinresistens.
Genetik och T2DM #
T2DM är en multifaktoriell sjukdom med en stark genetisk komponent, som framträder särskilt tydligt i familjekonstellationer. Risken för att syskon till en person med T2DM utvecklar sjukdomen är ungefär 2–3 gånger högre jämfört med familjer där ingen av syskonen har sjukdomen.
Om två syskon har T2DM stiger dock risken markant, upp till 30 gånger högre. Intressant nog är risken för att utveckla T2DM större om modern har sjukdomen, jämfört med om fadern är drabbad. Dessutom är risken för T2DM avsevärt högre hos individer med ett BMI på ≥30 eller ett förhöjt fastande blodsockervärde på över 5,5 mmol/l.
Att identifiera de genetiska faktorer som ligger bakom komplexa polygena sjukdomar som T2DM har länge varit en utmaning. Ett genombrott skedde 2007 med introduktionen av genomomfattande associationsstudier (GWAS), där vanliga genetiska varianter associerade med T2DM identifierades.
En av de mest framträdande upptäckterna var en enkel nukleotidpolymorfism (SNP) i TCF7L2, vilken visade den starkaste associationen med T2DM. Därefter har SNP-varianter i andra gener, såsom SLC30A8, FTO, CDKAL1, CDKN2A, CDKN2B, HHEX, IGF2BP2 och GCKR, också kopplats till sjukdomen. Sedan dess har listan över genetiska loci associerade med T2DM expanderat till över 300.
Majoriteten av dessa varianter finns i introniska regioner, vilket gör det mer korrekt att tala om genetiska loci snarare än specifika gener. Mekanismerna genom vilka dessa loci påverkar risken för T2DM är ofta svåra att klargöra. Ett fåtal exon varianter har dock visats påverka genfunktionen.
Exempel inkluderar SLC30A8, som kodar för en zinktransportör som krävs för insulinlagring, och KCNJ11, som kodar för en ATP-beroende kaliumkanal. Introner kan påverka genuttrycket av närliggande gener (i cis) eller avlägsna gener (i trans), även om detta hittills bara har bekräftats för ett fåtal gener, såsom en variant i MTNR1B, som kodar för en melatoninreceptor. Studier av odlade humana β-celler som bär riskalleler visar försämrad funktion och överlevnad av dessa celler.
Utmaningar i att förstå genetiska mekanismer
Trots betydande framsteg har förståelsen för mekanismerna bakom dessa genetiska varianter varit begränsad, och djurmodeller har inte alltid varit överensstämmande med humanfynd. Till exempel skyddar en mutation i den mänskliga SLC30A8-genen mot T2DM, medan samma mutation i möss predisponerar för sjukdomen.
Däremot har andra djurmodeller gett mer insikt. I en polygen modell för T2DM, Goto-Kakizaki-råttan, kunde en mutation i ADRA2A-genen kopplas till försämrad insulinsekretion. Överuttryck av α2A-adrenerga receptorer i β-celler förklarade denna defekt, och behandling med receptornedreglerande läkemedel förbättrade insulinutsöndringen både i humana och animaliska modeller.
Begränsningar och framtida forskning
De genetiska varianter som hittills identifierats har endast blygsamma effekter, vilket ökar risken för T2DM med cirka 10–20 %. Majoriteten av befolkningen, inklusive de utan diabetes, bär på dessa riskalleler, och deras genomsnittliga frekvens är 54 %.
Ändå har dessa varianter bidragit till nya insikter om patogenesen av T2DM. En prospektiv studie med cirka 2 700 individer visade att personer med hög genetisk risk (≥12 riskalleler) oftare utvecklade T2DM på grund av oförmåga att kompensera för den ökande insulinresistens som kom med ökad vikt. Däremot kunde individer med låg genetisk risk (≤8 riskalleler) bättre bibehålla insulinsekretionen trots liknande metabola utmaningar.
Trots framstegen förklarar de nuvarande genetiska varianterna endast 15 % av T2DM:s ärftlighet. Möjliga förklaringar inkluderar sjukdomens heterogenitet, gen-miljöinteraktioner och epigenetiska mekanismer som DNA-metylering och kromatinmodifieringar. Vissa varianter, såsom de i KCNQ1, visar starka föräldraursprungseffekter. Genen är metylerad och imprintad under fosterlivet men inte i vuxen ålder när den ärvts från modern.
Mitokondriell dysfunktion #
Mitokondriell dysfunktion har observerats i flera vävnader, inklusive lever, skelettmuskulatur, fettvävnad och hjärnan (inklusive hypotalamus), hos både djurmodeller och människor med fetma, T2DM och metabola syndromet. Detta fenomen beror på en kombination av reducerad mitokondrietäthet och nedsatt mitokondriell funktion, vilket i sin tur är en konsekvens av störd uttrycksnivå av olika komponenter i det oxidativa fosforyleringssystemet.
Den förändrade mitokondriella funktionen bidrar till insulinresistens på flera sätt. I fettvävnad har mitokondriell dysfunktion kopplats till försämrad sekretion av adiponektin, ett kraftfullt insulin-sensibiliserande adipokin. I andra vävnader, såsom lever och muskel, har mitokondriell dysfunktion föreslagits öka nivåerna av reaktiva syrearter (ROS), vilka aktiverar redoxkänsliga serinkinaser som fosforylerar IRS-proteiner och därmed orsakar insulinresistens.
Huruvida mitokondriell dysfunktion är en orsak eller en följd av insulinresistens är fortfarande föremål för diskussion. Dock, med tanke på den centrala roll som ektopisk lipidinlagring har i utvecklingen av insulinresistens, är mitokondriell dysfunktion—som ofta är förknippad med minskad fettsyreoxidation i mitokondrier—åtminstone en viktig förvärrande faktor i denna process.


